

ORF- und Gesamtgenomsequenzierung des porcinen reproduktiven und respiratorischen Syndrom-Virus (PRRSV) in Routineproben führen zum Nachweis eines rekombinanten Virusstammes
Berliner und Münchener Tierärztliche Wochenschrift 133
DOI: 10.2376/1439-0299-2020-19
© Schlütersche Verlagsgesellschaft mbH & Co. KG. 2020
Publiziert: 10/2020
Summary
Porcine reproductive and respiratory syndrome virus (PRRSV) causes one of the economically most important diseases in pig farming. In view of the particularly high level of genetic variation of this virus, molecular epidemiological methods are very well suited for the characterisation of virus strains. In our study, we present the results of PRRSV testing using RT-qPCR and following ORF5, ORF4–ORF6 and whole genome sequencing (WGS) in two pig farms, connected by purchase and both suffering from PRRSV outbreaks. Analyses based on ORF5 sequences detected only a field virus strain in farm A, despite vaccination with a modified live vaccine (MLV). In farm B, the MLV used in farm A and a field virus strain could be determined. However, the ORF4–ORF6 sequences confirmed the results from farm A, but revealed two different field virus strains in farm B. This phenomenon could be resolved using next generation sequencing (NGS) technologies for WGS analyses of two representative virus strains originating from farm B. WGS revealed two closely related field virus strains with a nucleotide sequence identity of 97.72%. One strain was a recombinant PRRSV strain, apparently created from the MLV used in farm A and a field virus strain. The fact that only the ORF5 region originated from the MLV explains the differences in the sequences of ORF5 compared to ORF4–ORF6 and WGS. Our study shows that ORF-based molecular characterisation of PRRSV strains might be deceptive due to mosaic recombination events requiring WGS to accurately reveal the molecular relationships between virus strains.
Zusammenfassung
Das porcine reproduktive und respiratorische Syndrom-Virus (PRRSV) verursacht eine der wirtschaftlich wichtigsten Krankheiten in der Schweinehaltung. Im Hinblick auf die besonders große genetische Varianz des Virus eignen sich molekular-epidemiologische Methoden zur Charakterisierung von Virusstämmen sehr gut. In unserer Studie stellen wir die Ergebnisse der PRRSV-Testung mittels RT-qPCR und anschließender Sequenzierung von ORF5 und ORF4–ORF6 sowie Gesamtgenomsequenzierung (WGS) in zwei durch Kauf von Tieren in Kontakt stehenden Schweinebetrieben mit PRRSV-Ausbrüchen vor. Bei ausschließlicher Verwendung von ORF5-Sequenzen konnte in Betrieb A trotz Impfung mit einem modifizierten Lebendimpfstoff (MLV) nur ein Feldstamm nachgewiesen werden. Im Gegensatz hierzu gelang in Betrieb B der Nachweis des in Betrieb A eingesetzten MLV sowie eines Feldstammes. Die ORF4- bis ORF6-Sequenzen bestätigten die Ergebnisse aus Betrieb A, ergaben jedoch in Betrieb B zwei unterschiedliche Feldstämme. Dieses Phänomen konnte mithilfe einer Gesamtgenomanalyse anhand der Next Generation Sequencing(NGS)-Technologie von zwei repräsentativen Virusstämmen aufgeklärt werden. Anhand der WGS konnten zwei eng verwandte Feldviren mit einer Nukleotidsequenzidentität von 97,72 % nachgewiesen werden. Ein Virusisolat stellte sich dabei als ein rekombinanter Virusstamm heraus, offensichtlich zusammengesetzt aus dem in Betrieb A verwendeten MLV und einem Feldvirus. Da nur die ORF5-Region aus dem MLV stammte, erklärt dies, weshalb sich die Sequenzen von ORF5 gegenüber denen von ORF4–ORF6 und WGS signifikant unterschieden. Die Ergebnisse zeigen, dass eine ORF-basierte molekulare Charakterisierung von PRRSV-Stämmen aufgrund von unterschiedlich lokalisierten Rekombinationsereignissen irreführend sein kann. Nur die Sequenzierung des gesamten Genoms kann die korrekte molekulare Verwandtschaft zwischen Virusstämmen aufdecken.
Introduction
Porcine respiratory and reproductive syndrome (PRRS) is one of the economically most important diseases in global pig production, and is caused by the PRRS virus (PRRSV). Annual losses caused by PRRS in the United States are estimated to be $664 million (Holtkamp et al. 2013). In Germany, PRRS was detected for the first time in November 1990 (Drew 1995). Outbreaks in Baden-Württemberg (a federal state of Germany) in 2012–2013 cumulated in costs up to €135,000 per farm and outbreak (Richter et al. 2014).
PRRSV is a 45–60 nm in diameter enveloped RNA virus with a positive-sense, single stranded RNA genome of around 15 kbp with at least 10 open reading frames (ORFs) (Dokland 2010). Belonging to the order of Nidovirales, family Arteriviridae, PRRSV is nowadays classified into the genus Betaarterivirus with the two different subgenera: Eurpobartevirus with the species Betaarterivirus suid1 (PRRSV-1, originating from Europe) and subgenus Ampobarterivirus, with the species Betaarterivirus suid 2 (PRRSV-2, originating from North America) (ICTV, 2018). Like other RNA viruses, PRRSV has a high genetic variability (Stadejek et al. 2006) with a high mutation rate (Hanada et al. 2005), whereby the ORF-5 region seems to be one of the most variable regions of the genome (Martin-Valls et al. 2014; Shi et al. 2010a, b). This variability is caused by a relatively poor fidelity of the RNA-polymerase (Castro et al. 2005) and selective pressures by the immune system (Mateu et al. 2006). The ORF 5 encodes the Major Glycoprotein 5 (Dokland et al. 2010) and this Glycoprotein is part of the envelope, participates in viral attachment to cells, contains a neutralisation epitope and is therefore a target for the immune system (Mateu et al. 2006).
To control acute PRRSV outbreaks, the use of modified live vaccines (MLV) can be effective (Charerntantanakul, 2012). The correct use of live vaccines combined with further measures such as early diagnosis and monitoring, high level of biosecurity, strict hygiene, and an optimized herd management, promises high stock protection. As a result, a reduction in clinical symptoms as well as viral shedding due to an ongoing repression of the field virus can be expected (Charerntantanakul, 2012; Pileri and Mateu, 2016; Rathkjen, Dall, 2017). However, there is also the risk of a recombination event between field and vaccine viruses by using MLVs during an acute PRRSV outbreak (Charerntantanakul, 2012; Eclercy et al. 2019; Montaner-Tarbes et al. 2019). Homologous recombination of two field viruses (Martin-Valls et al. 2014; Yuan et al. 1999), of a field and vaccine viruses and even of two vaccine viruses have been described in previous studies (Burgara-Estrella et al. 2014, Li et al. 2009; Steinrigl, 2018; CVMP, 2019; Eclercy et al. 2019; Marton et al. 2019). PRRSV type 1 vaccine strains leading to a recombinant strain that has been associated with clinical signs of disease in PRRS-naive herds in Denmark has recently prompted the Committee for Medicinal Products for Veterinary Use (CVMP) to make recommendations. These concern the use of live attenuated PRRSV vaccines and reporting of any suspected adverse incidents and sequence data which indicate recombination events of vaccine strains with each other or wild types (CVMP, 2019).
Top Job:
In our case report, we present the results of PRRS outbreaks in two farms which came into contact through the purchase of animals. Our aim is to elucidate the relationship of the PRRSV strains that were responsible for these PRRS outbreaks. The comparison of sequence data obtained from Sanger sequencing of ORF5 and ORF4–ORF6 attracted our attention due to the different results. To resolve this phenomenon, we decided to use next generation sequencing (NGS) technologies for whole genome analyses.
Material and Methods
Origin of samples
Farm A – piglet production
In September 2017, in a piglet production farm with 400 sows, nearly all suckling piglets showed suddenly fever, anaemia, coughing and poor suckling. Tissue samples from the lung were taken from an euthanised piglet (piglet A) and blood samples from ten suckling piglets pooled in two samples (serum pool 1 and 2) were tested by PRRSV reverse transcription real-time PCR (RT-qPCR) followed by ORF4–ORF6 Sanger sequencing. The routine vaccination scheme for the suckling piglets on this farm consisted of a PRRSV-1 MLV (named MLV II) vaccine and a combination vaccine against Mycoplasma hyopneumoniae and PCV-2. The sows were vaccinated every four months with the same PRRSV-1 MLV and a combination vaccine against Parvovirus und erysipelas.
Farm B – piglet rearing farm
At the same time, the piglet rearing farm B which had received 4-week-old piglets with an average weight of 8 kg from farm A, reported greater differences in the growth of the piglets and an increase in the number of runts suffering from coughing and sneezing. These symptoms were evident in all age groups. Fifteen blood samples tested in three serum pools (serum pool 3, 4 and 5) and lung tissue (piglet B) were taken from piglets with typical symptoms to test for PRRSV using RT-qPCR. Sequencing of ORF4–ORF6 was performed for PRRSV from lung tissue of piglet B and from serum pool 3. Five months later, in February 2018, piglets again showed slight coughing, sneezing and sporadic mild conjunctivitis. Therefore, three serum pool samples from ten piglets (serum pool 6, 7 and 8) were tested by PRRSV RT-qPCR. Subsequently, serum pool 7 and serum pool 8 were examined by Sanger sequencing of the ORF4–ORF6 region and NGS to obtain whole genome sequence data. Due to the vaccination of the pigs with a PRRS MLV in farm A, no further vaccination against PRRSV was administered to the pigs.
RNA isolation and PRRSV RT-qPCR
Viral RNA for RT-qPCR was isolated from blood samples and lung tissue using the MagMAX Pathogen RNA/DNA Kit (Thermo Fisher Scientific, Waltham, USA) following the manufacturer´s instructions.
For conventional PCR and following sequencing, RNA was isolated from lung tissue as described before (Frey et al. 2017) using a TRIzol® reagent (Thermo Fisher Scientific, Waltham, USA) for pre-treatment and the RNeasy Mini Kit (Qiagen, Hilden, Germany). Briefly, 50 mg organ sample was initially transferred into a grinding tube and homogenised in 1 ml TRIzol® reagent at 5000 rpm / 5 sec / 1 cycle using the Precess 24 (Bio-Rad Laboratories, Hercules/ Kalifornien, USA). After centrifugation (1 minute, 16 000 x g) and incubation (5 min at room temperature), 200 μl chloroform (chloroform minimum 99%, Sigma-Aldrich, Taufkirchen, Germany) was added; the mixture was vigorously shaken, incubated for 10 min at room temperature and centrifuged (10 min, 4°C, 16 000 x g). The upper aqueous phase (500−600 μl) and 600 μl of 70% ethanol (Carl Roth, Karlsruhe, Germany) were thoroughly mixed and transferred into an RNeasy Mini Spin column. The following purification steps were performed with the RNeasy Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions.
RNA for NGS analysis was isolated from 200 µl serum using the MagMAX™ CORE Nucleic Acid Purification Kit and a KingFisher Flex device according to the manufacturer´s instructions (Thermo Fisher Scientific, Waltham, USA). Each sample was prepared twice, with and without a DNase treatment. For the DNase digestion the script of the manufacturer was extended. After the second washing step the beads were incubated for 15 min at RT with the DNase solution (10 µl DNase Stock I + 70 µl RDD; Qiagen RNase free DNase Set; Qiagen, Hilden, Germany). After two further washing steps the beads were eluted at 65°C in 90 µl elution buffer.
For PRRSV screening and species identification (PRRSV-1, EU and PRRSV-2, US and HP) the commercially available virotype PRRSV RT-qPCR Kit (Indical Bioscience, Leipzig, Germany) was used according to the manufacturer´s instructions on a CFX96 TouchTM Real-Time PCR Detection System (Bio-Rad Laboratories, Hercules/ Kalifornien, USA).
Conventional RT-PCR
In preparation for the Sanger sequencing, a conventional PCR with following gel electrophoresis was performed. As a result of validation and optimization processes, two different protocols were used for the PCR for blood samples and lung tissue.
For blood samples, the isolated RNA and the primers PRRS-ORF4-F (TGG TTT CTS AGG CGT TCG C) and PRRS-ORF6-R (GGG CAG GGG CCA GAA TGT) (Frey et al. 2017) were added to the SuperScriptTM III One-Step RT-PCR System with PlatinumTM Taq (Thermo Fisher Scientific, Waltham, USA) and the reverse transcription PCR was performed according to the manufacturer´s instructions with slight modifications: the cycler programme steps consisted of 15 min at 50°C followed by 2 min at 95°C for cDNA synthesis and pre-denaturation, respectively. These steps were followed by 45 cycles of a) 15 sec at 95°C, b) 30 sec at 57°C, c) 90 sec at 72°C, finalised by 5 min at 72°C. The master mix consisted of 12.5 µl 2x Reaction Mix, 1 µl Taq-Mix, 1 µl MgSO4, 2 µl forward primer (10 µM), 2 µl reverse primer (10 µM), 1.5 µl DEPC water, to which 5 µl template RNA per sample was added.
Using lung tissue, a three-step PCR was performed as published previously (Frey et al. 2017). The isolated RNA and anchored-oligo (dT)18 primer were added to the PCR reagents provided by Roche (Transcriptor High Fidelity cDNA Synthesis Kit, Roche, Mannheim, Germany) and the RT-PCR was performed according to the manufacturer´s instructions. In the third step, the PCR was carried out using the FastStart High Fidelity PCR System, dNTPack (Roche, Mannheim, Germany) according to the manufacturer´s protocol, also using the PRRS-ORF4-F and PRRS-ORF6-R primers as mentioned before.
DNA generated by RT-PCR was analysed in 2% agarose gel. Gel electrophoresis and visualisation by staining with peq-GREEN (VWR, Erlangen, Germany) revealed a single band with a size of around 1,450 bp. The PCR product was then purified using the Mag-Bind® Total Pure NGS Kit (Omega Bio-tek, Norcorss, Georgia, USA) run on a KingFisher Flex device (Thermo Fisher Scientific, Waltham, USA) for subsequent DNA Sanger sequencing on demand (Microsynth, Balgach, Switzerland).
Sequencing
Samples yielding Cq values 22 in the RT-qPCR were forwarded for sequencing of the ORF4–ORF6 region on demand using Sanger sequencing (Frey et al. 2017). The amplified DNA strand was sequenced in both the forward and reverse direction for confirmation.
The whole genome sequencing of the serum pool samples 7 and 8 was performed on demand using NGS technology (CeGaT, Tübingen, Germany). Both samples were chosen based on the results of the previous Sanger sequencing. As quality controls, the RNA content, concentration and RIN were determined using Qubit RNA (Thermo Fisher Scientific, Waltham, USA) and the Bioanalyzer RNA Pico (Agilent, Santa Clara, USA), respectively. For library preparation with the NuGen Trio RNA-Seq Library Preparation Kit (Tecan Genomics, Redwood City, USA), an amount of 0.8–2.5 ng RNA was used. Sequencing was carried out by the MiSeq 300 cycle v2 Sequencing Kit (Illumina, San Diego, USA) with 2 x 150 bp, yielding QC-values of 89.56% Q30 (Supplementary Table S1).
Analysis of sequence data
The program Geneious Prime 2019.1.1 (Biomatters, Auckland, New Zealand) was used for further processing and evaluation of the obtained sequence data. For the sequences obtained by Sanger sequencing, the forward and the reverse sequences were merged into a common consensus sequence and then aligned with other sequences from this case and from the CVUAS PRRSV database (multiple alignment with other regional field isolates, reference strains and vaccine strains, data not shown) (Frey et al. 2017). For the alignment, the “Geneious Alignment” (program Geneious Prime 2019.1.1, Biomatters, Auckland, New Zealand) was chosen; performing a global alignment with free end gaps, cost Matrix: Identity (1.0/0.0), Gap open penalty 12 and gap extension penalty 3.
For bioinformatics analysis of the NGS data, the Illumina bcl2fastq (2.19) (Illumina, San Diego, USA) was used for the de-multiplexing of the reads. Adapters were trimmed using Skewer (Version 0.2.2) (Jiang et al. 2014). Further quality trimming of the reads was not performed. The FASTQC (Brahaman Bioinformatics, version 0.11.5-cegat) (CeGaT, Tübingen, Germany) was used for analyses of the FASTQ files. The FASTQ-files of the NGS were imported into the programme Geneious Prime 2019.1.1. After pairing, duplicates were removed by the Plug-In Dedupe Duplicate Read Remover (Version 38.37 by Brian Bushnell) using a Kmer seed length of 31, zero maximum edits and zero maximum substitutions. The quality trimming was performed by the Geneious programme (error probability limit: 0.01; filtering post-trim > 50). The following mapping was carried out by the Geneious mapper programme, using up to five iterations for fine-tuning. The previously trimmed reads were mapped to the Sscrofa11.1 (GCF_0000003025.6) sequence to identify reads belonging to Sus scrofa. (Supplementary Table 1). All non-mapped reads were mapped on a database of 775 PRRSV whole genome sequences (NCBI, GenBank, Bethesda MD, USA; download: 19.06.2018; Supplementary Table S2).
The sequence with the highest sequence coverage was set as reference sequence and the mapping was repeated against this one sequence (KT033457 for all four sequences), leading to a consensus sequence (Zhang et al. 2017). The consensus sequences of the preparations with and without DNase treatment were finally aligned, generating the final viral whole genome consensus sequence. A de-novo assembly of the reads was performed by using different software tools (Supplementary Document S3).
Because ORF5 is the preferred region for characterisation of the PRRSV genome by sequencing due to its high genetic variability (Dortman et al. 2019), we extracted the ORF5 (604 nt) from the ORF4–ORF6 sequences. Then we aligned the sequences of all virus strains of this case with the MLV II, on the one hand only based on ORF5 and on the other hand based on the longer sequences of ORF4-ORF6. The results of the alignments, especially the nucleotide sequence identity between the virus strains and MLV, were compared. The interpretation of the results based on the assumption, that virus strains with a nucleotide sequence identity of more than 97% evolved from one common origin (Cortey et al. 2017). Therefore, the virus strains were classified as MLV derived strains or as one or more related or unrelated field strains.
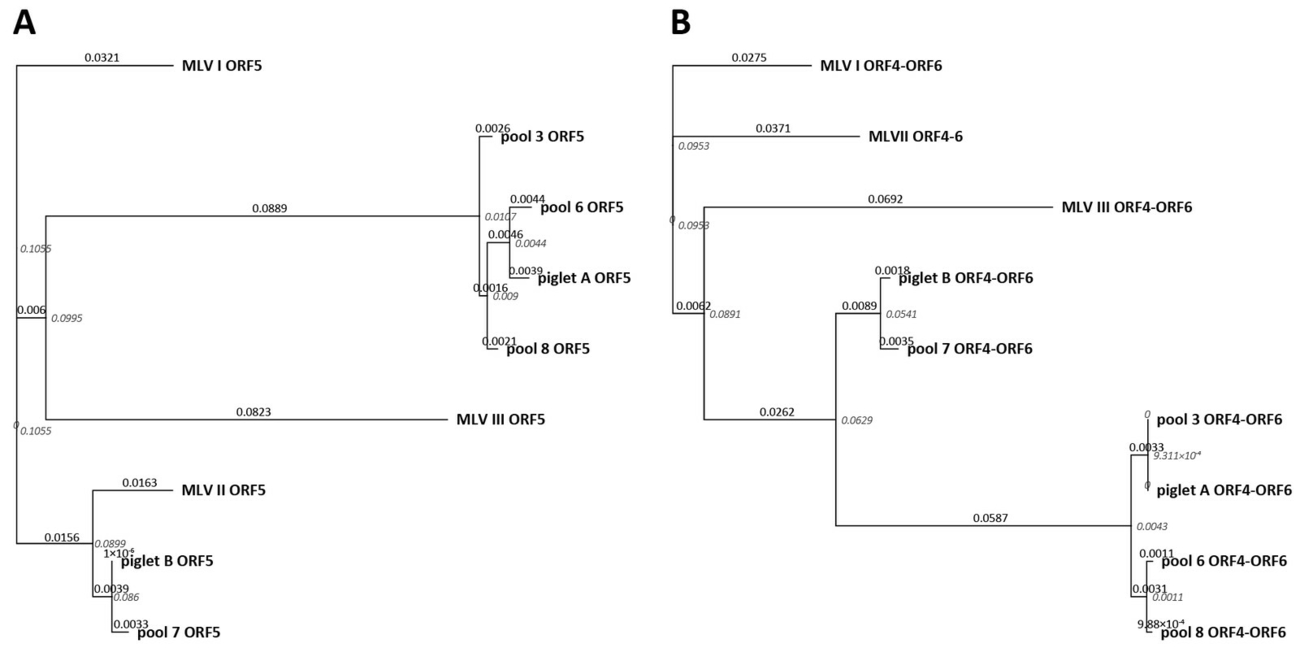
In addition, phylogenetic trees were constructed with the Geneious Tree Builder (Biomatters, Auckland, New Zealand) using Tamura-Nei as the Genetic Distance Model and Neighbor-Joining as the tree build method. The phylogenetic trees base on the alignment of ORF5 and ORF4-ORF6 sequences of this case and three modified live vaccines (MLV I, MLV II, MLV III) (Frey et al. 2017).
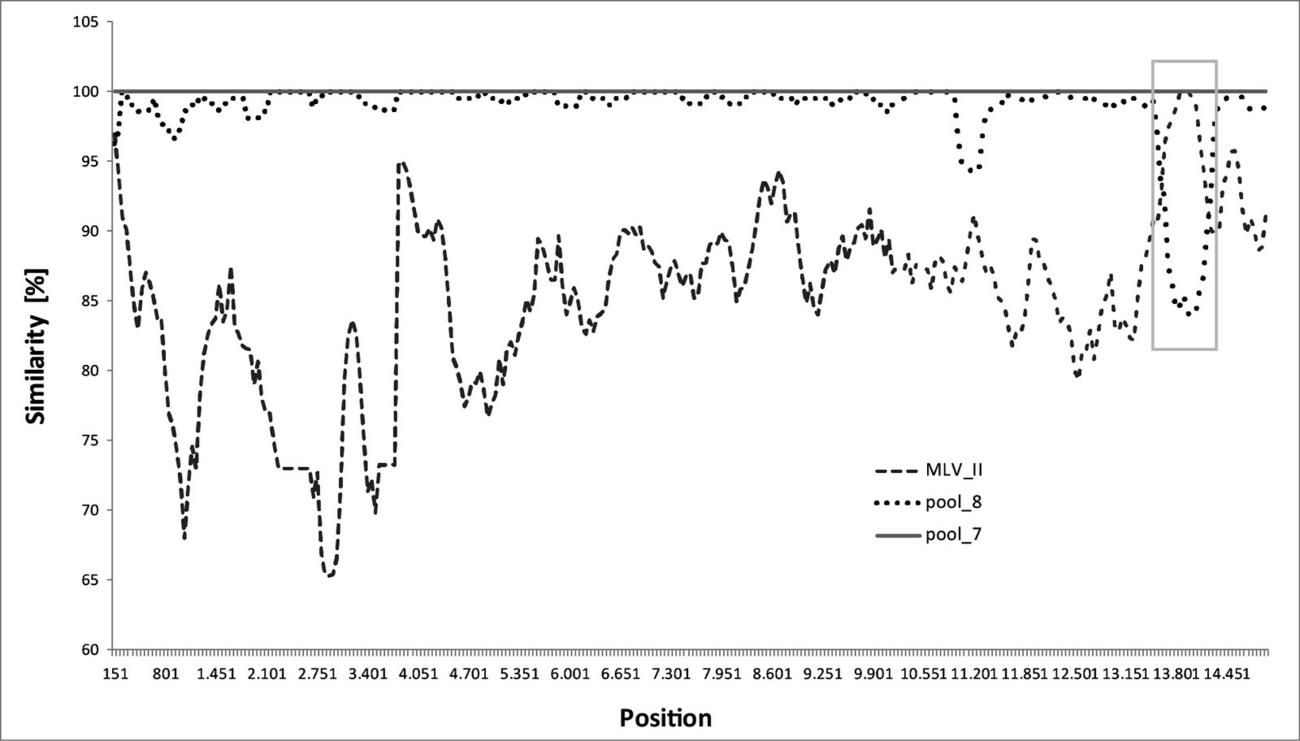
The recombination analysis was performed using RDP4, a recombination detection programme that implements an extensive array of methods for detecting and visualising recombination in virus genome sequence alignments (Martin et al. 2015). As second tool, a similarity plot was generated with help of SlimPlot (Lole et al. 1999), using serum pool 7 as query sequence and MLV II and serum pool 8 as comparison sequences. In both plots, recombination sites are identified on crossover regions and breakpoints are determined.
Results
RT-qPCR
In September 2017, nearly all suckling piglets in a piglet production farm suddenly showed fever, anaemia, coughing and poor suckling. Tissue and blood samples from this farm, as well as from a second affected holding, were tested by PRRSV RT-qPCR followed by ORF4–ORF6 Sanger sequencing of selected samples. Subsequently, two serum pools were examined by NGS to obtain WGS data.
In farm A, a high viral load of PRRSV-1 could be detected in the lung tissue of piglet A and in one of the tested serum pools by RT-qPCR. In farm B, all samples taken at two different times tested positively for PRRSV-1 by RT-qPCR with variable viral load (Table 1).
Sequencing
The results of the alignments based on ORF5 and based on ORF4-ORF6 were compared. Sequences of the PRRSV strain obtained from piglet A (farm A) revealed high nucleotide sequence identity values (> 98.68%) to PRRSV sequences retrieved from the serum pools 3, 6, and 8 (farm B), but low nucleotide sequence identities to the vaccine strain MLV II, piglet B and serum pool 7 (both farm B) ( 93.06%). This applies for the ORF5 and ORF4–ORF6 sequences (Table 2). This means that the administered vaccine strain MLV II in farm A could not be detected, but a field strain in piglet A that had also been detected in farm B in three serum pools (serum pool 3, 6 and 8) could be detected.
In farm B, the PRRSV strain detected in serum pool 7 showed a high nucleotide sequence identity to the strain found in piglet B based on ORF 5 and ORF4–ORF6 sequencing (99.67% and 99.47%, respectively). The nucleotide sequence identities of serum pool 7 and piglet B to the vaccine strain MLV II was very high based on ORF5 (98.01% and 97.68%, respectively), but yielded low nucleotide sequence identity values (94.02% and 93.7%, respectively) when using ORF4–ORF6 sequences (Table 2). The findings based on ORF5 sequencing suggest the presence of a MLV II vaccine derived strain in farm B; however, confirmation by ORF4–ORF6 sequencing failed. In fact, the nucleotide sequence identity values of the ORF4–ORF6 sequencing suggest the presence of two different field virus strains in farm B.
A phylogeny analysis based on ORF 5 and ORF4–ORF6 sequences was performed to investigate the relationship of the different isolates in this study (Figure 1B). Within the ORF5 based phylogenetic trees, the sequences of piglet B and serum pool 7 form a cluster containing MLV II (Figure 1A). However, if the analysis is based on ORF4–ORF6 sequences, the piglet B/pool 7 cluster is intercalated between the MLV and the field strain cluster (Figure 1B).
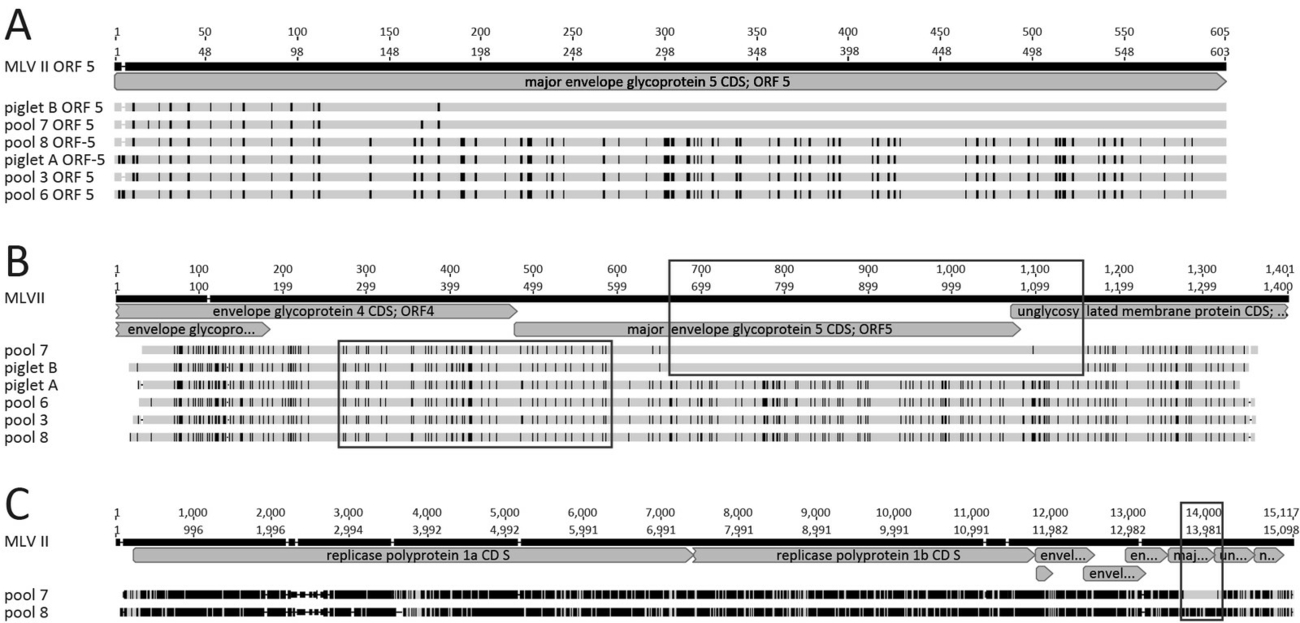
The alignments of the ORF5 sequences show a clear difference in the distribution of nucleotides differing from MLV II (reference) between piglet B and serum pool 7 on the one hand, and piglet A and the serum pools 3, 6, and 8, on the other hand (Figure 2A). Expanding the examined genome section from ORF5 to ORF4–ORF6 for more detailed analyses, it becomes evident that the nucleotide differences of the PRRSV strains originating from piglet B and serum pool 7 compared to piglet A and the serum pools 3, 6, and 8 are localized especially towards the 3’ region of ORF5 (Figure 2B). However, approximately 300 nucleotides within the sequences of ORF4 and of the directly adjacent ORF5 proved to be very similar (Figure 2B), suggesting a possible recombination event.
Due to these inconsistent results obtained by ORF5 and ORF4–ORF6 sequencing, a NGS of the whole genome was performed on serum pool 7 representing the vaccine strain MLV II in the ORF5 region, and serum pool 8 representing the field strain cluster for comparative purposes. With a sequence identity of 97.72% over the whole sequence, these two viral strains could be considered as field virus strains of one common origin and clearly separated from the vaccine strain MLV II, yielding a nucleotide sequence identity value of only 83.43% and 85%, respectively. A closer look at the alignment of whole genome sequences shows that only a part the ORF5 sequence of the PRRSV strain obtained from serum pool 7 shows a high nucleotide sequence identity to the MLV II strain, while the other regions of the genome match with serum pool 8 but not with MLV II (Figure 2C).
In further evaluations of the WGS data, the recombination event could be localized exclusively to the genome nucleotide positions 13,648 to 14,186 by recombination analyses using the programmes RDP4 (recombination detection program) (Martin et al. 2015) (data not shown) and to the genome nucleotide positions 13,600 to 14,200 using SlimPlot (Lole et al. 1999). Both analysis procedures revealed that the PRRSV strain detected in serum pool 7 shows strong evidence of being a recombinant virus strain derived from serum pool 8 as major parent, and MLV II as minor parent (Figure 3). The recombination site comprises 538 nt using RDP4 or 600 nt using SlimPlot, respectively.
Validation of the NGS data
The DNase treatment was primary performed to determine any influences on the results. As expected, it had no effect, but this completely independently generated set of data could be used as duplicate and merged with the correspondent sequences without DNase treatment to a consensus sequence (data not shown). By this way we could increase the quality of the data and assure a confirmed and genome wide high read coverage. A de novo assembly was performed by Tadpole (http://seqanswers.com/forums/showthread.php?t=61445). An alignment of the reference sequence MT857222 with the de novo assembled contigs of pool 7 showed a covering of 75.4% of the genome, an alignment of the reference sequence MT857223 with the de novo assembled contigs of pool 8 showed a covering of 97.2%. The pairwise nucleotide identity was very high in both alignments (pool 7 99.7% and pool 8 98.4%) (Supplementary Document S3).
Furthermore, the validity of the sequences generated by NGS could be confirmed by an alignment with the sequences achieved by Sanger sequencing (nucleotide sequence identity of 100% for pool 7 and pool 8; data not shown). Even though only a short part of the whole genome could be verified in this way, it was sufficient to prove that the entire recombination region was not a bioinformatics artefact from mapping.
Discussion
Control of PRRS is important due to the enormous economic losses caused by this viral disease, but is challenging due to the high genetic variability of the virus (Frey et al. 2017; Hanada et al. 2005; Stadejek et al. 2006). Furthermore, virus variability is enhanced by genetic recombination with modified live vaccines (MLV) which are used as a very effective preventive tool (Eclercy et al. 2019). These features of the PRRSV make molecular epidemiology for characterisation of PRRSV strains very valuably. The results of molecular epidemiology allow taking specific measures concerning biosecurity, hygiene, and vaccine management to control the PRRSV situation (Dortmans et al. 2019; Frey et al. 2017).
In our case study, we could show the importance of sequencing and the impact of sequence data of the PRRSV genome on the interpretation of results in routine diagnostics. Our results show that conclusions based on partial sequencing should be drawn cautiously and depend on the available sequence data volume. In the case presented, analysing only the ORF5 sequences leads to the contradictory interpretation that the PRRSV detected in piglet B and serum pool 7 in farm B is a vaccine virus (nucleotide sequence identity 98.01% and 97.68% to MLV II, respectively), although no vaccination had been administered. As a consequence of this assessment, it could be concluded that the MLV vaccine strain had established a clinically apparent PRRSV infection in farm B, even though no vaccine measures had been taken. This would be of critical concern to the pharmacological safety of modified live vaccines. In contrast, evaluation of the ORF4–ORF6 sequences reveals two circulating field viruses in piglet B and serum pool 7, respectively (nucleotide sequence identity of 94.02% and 93.7% to MLVII, respectively). This scenario would result in tracing the source of the PRRSV field strains to adapt and improve biosecurity measurements (Cortey et al. 2017; Dortmans et al. 2019).
Inconsistent results of the nucleotide sequence identity values obtained by ORF5 and ORF4–ORF6 sequencing and the results of comparative alignments of the sequences of these ORF regions led to the suspicion that a recombination event had occurred. Therefore, we decided to sequence the whole PRRSV genome. Analysing the whole genome data and performing a recombination analysis revealed that the PRRSV detected in serum pool 7 proved to be neither a vaccine virus nor a second field virus strain, but a recombinant virus strain with the field virus as major and the MLVII vaccine virus strain as minor parent. The recombination event that had occurred exclusively in the ORF5 sequence which represents the most commonly used sequence for molecular epidemiology (Martin-Valls et al. 2014; Shi et al. 2010a, b), masked the true presence of a field virus/MLV recombinant PRRSV-1 strain when focusing only on evaluation of the ORF5 sequences. There are several studies reporting on recombinant field virus strains that had been characterised as vaccine virus strains based on ORF5 sequencing. However, using WGS and recombination analysis, only parts of the ORF5 region could be attributed to sequences of a vaccine virus strain, whereas other parts of the genome apparently originated from a field virus strain as a result of recombination events (Marton et al. 2019; Steinrigl et al. 2018). These findings support the assumption that ORF5 is a hotspot of recombination (Li et al. 2009; Murthaugh et al. 2001; Shi et al, 2010a, b) but recombination events can be found also in other genome regions, with or without involvement of ORF5 (Eclercy et al. 2019; Marton et al. 2019;). For characterisation of the PRRSV based on WGS data, it is necessary to make the whole image of the genetic mosaic visible for accurate epidemiological studies (Cortey et al. 2017). It has been shown that partial sequences lead to different interpretation regarding the relationship of viruses, independent of the use of a highly variable region like ORF5 or a more stable region like ORF7 (Dortmans et al. 2019; Martin-Valls et al. 2014). Therefore, creating phylogenetic trees of the same virus strains based on different genome sequences might differ significantly (Martin-Valls et al. 2014; Stadejek, 2012). Hence, each ORF-based sequencing result might be misinterpreted, but in-depth studies using WGS may help to solve relevant issues (Cortey et al. 2017). Currently, the use of WGS data in routine diagnostics is limited due to the current high price and large amount of data generated for sophisticated analyses and comparative evaluations (Cortey et al. 2017). However, it can be expected that prices are dropping, and large amounts of data generated by WGS will be processed more rapidly in the near future, superseding partial sequencing. The benefits of WGS outweigh the costs significantly, especially when incorrect conclusions in diagnosis, therapy and epidemiology drawn from misinterpretation due to partial sequences can be avoided (Cortey et al. 2017, Dortmans et al. 2019).
The clinical relevance of this hypothetical recombination event remains unclear. Cortey et al. (2017) advises to be very careful with incorrect conclusions based on mutation analysis only by NGS. The differentiation between biological significant mutations regarding adaption or mutation or stochastic mutations should be determined in in vitro and in vivo experiments in order to conclude biological properties. Up to now, consistent genetic signatures of virulence were not known (van Geelen et al, 2018).
Another important aspect that emerged from our study is that multiple PRRSV strains occurred in farm B at the same time and over a longer period. This phenomenon of circulating and re-circulating of identical PRRS strains in swine herds had also been previously described by Larochelle et al. (2003) and recently by Torrents et al. (2019). Consequently, single samples are not sufficient for a sound assessment of the PRRSV situation in a farm due to super- and coinfection, mutation and recombination (Larochelle et al. 2003). In general, recombination can occur if one cell is coinfected by two or more virus strains at the same time and can be considered as basic process in the development of diversity of RNA viruses (Simon-Loriere and Holmes, 2011), but the exact mechanisms for arterivirus homologous and nonhomolougus recombination are not completely understood (Kappes and Faaberg, 2015). Unfortunately, we could not clarify whether the recombinant virus strain had been created in the production farm A caused by using the MLV life vaccine, or later in the piglet rearing farm B. Nonetheless, the use of a modified live vaccine during an acute PRRSV outbreak usually favours the creation of recombinant viruses (Eclercy et al. 2019). Recombination events between field and vaccine viruses are quite often described in the literature (Eclercy et al. 2019, Marton et al. 2019; Steinrigel, 2019) and are one of the major safety issues regarding vaccine strategies (Montaner-Tarbes et al. 2019). This is a current issue that caused the Committee for Medicinal Products for Veterinary Use of the European Medicines Agency to release recommendations on the use of live attenuated PRRSV vaccines, due to clinical signs of disease in PRRSV-naive herds in Denmark being ascribed to recombination events of PRRSV type 1 vaccine strains (CVMP, 2019). Evaluation and collecting data obtained from WGS of recombinant PRRSV strains is therefore of particular relevance.
Beside the risk of reversion of a vaccine strain to a virulent PRRSV strain (Kappes and Faaberg, 2015; Montaner-Tarbes et al. 2019), recombination events might lead to viruses with increased pathogenicity (Montaner-Tarbes et al. 2019). Furthermore, recombination events are even considered to be the cause of reversion of circulating PRRSV-2 strains in China to highly pathogenic PRRSV-2 (HP-PRRSV) with increased fatality and a wide spread around China and Southeast Asia (Shi et al. 2013). Eclercy et al. (2019) also showed that the recombination of two modified live vaccine viruses had led to increased viral excretion and transmission compared to parental vaccine strains and a higher viral fitness with the increased risk of reversion to virulence due to genetic evolution. In Denmark, a recombination event in two live attenuated PRRSV type 1 vaccine strains also led to clinical signs of disease in PRRSV-naive herds (CVMT, 2019). In our case, the recombinant MLV II/field virus strain was also able to circulate simultaneously together with an “established” field virus over months. Indeed, all these findings support the demand for a careful use of MLV live vaccines for prevention and control of PRRSV in order to avoid recombination events and the consequent risk of evolution of novel, more pathogenic virus strains (Charerntantanakul, 2012; Eclercy et al. 2019; Kappes and Faaberg, 2015; Montaner-Tarbes, 2019).
Conclusion
Inconsistent sequencing results of single ORFs should be verified by in-depth WGS sequencing using NGS to achieve accurate molecular epidemiological interpretations. For phylogenetic analysis and detection of mosaic recombination events, full genome sequences are most appropriate for a comprehensive view of the PRRSV genome.
Furthermore, NGS provides the opportunity for a deep and open-view diagnostic method when sequencing of the metagenome including information on all viruses contained in a sample. A crucial issue to achieve these goals is the use of reliable methods for extraction of nucleic acids from different matrices, standardisation of data analyses and creation of comprehensive up-to-date reference datasets on an open access web-based genotyping platform.
Acknowledgment
The authors thank Agnes Richter and colleagues from the Schweinegesundheitsdienst Baden-Württemberg for the sample collection and the great collaboration.
We thank Julia Skrypski and Jasmin Stelzer for excellent performance of the RT-qPCR assays and comparison and alignments of nucleotide sequences.
We would also like to thank Pat Schreiter for the excellent creation of the graphics.
Conflict of interest
The authors declare that they have no conflict of interest.
Address for correspondence
Dr. Lisa Schneider-Bühl
Chemisches und Veterinäruntersuchungsamt Stuttgart
Schaflandstr. 3/3
70736 Fellbach
Germany
Lisa.Schneider-Buehl@cvuas.bwl.de
References
Burgara-Estrella A, Reséndiz-Sandoval M, Cortey M, Mateu E, Hernández J (2014): Temporal evolution and potential recombination events in PRRSV strains of Sonora Mexico. Vet Microbiol 174: 540–546.
Castro C, Arnold JJ, Cameron CE (2005): Incorporation fidelity of the viral RNA-dependent RNA polymerase: a kinetic, thermodynamic and structural perspective. Virus Res 107: 141–149.
Charerntantanakul W (2012): Porcine reproductive and respiratory syndrome virus vaccines: immunogenicity, efficacy and safety aspects. World J Virol 1: 23–30.
CVMP (Committee for Medicinal Products for Veterinary Use) of the European Medicines Agency (2019): CVMP recommendation on the use of live attenuated PRRSV vaccines. Press release EMA/CVMP/629092/2019. www.ema.europa.eu/en/news/committee-medicinal-products-veterinary-use-c….
Cortey M, Díaz I, Martín-Valls GE, Mateu (2017): Next-generation sequencing as a tool for the study of Porcine reproductive and respiratory syndrome virus (PRRSV) macro- and micro-molecular epidemiology. Vet Microbiol 209: 5–12.
Dokland T (2010): The structural biology of PRRSV. Virus Res 154: 86–97.
Dortmans JCFM, Buter GJ, Dijkman R, Houben M, Duinhof TF (2019): Molecular characterization of type 1 porcine reproductive and respiratory syndrome viruses (PRRSV) isolated in the Netherlands from 2014 to 2016. PLoS ONE 14: e0218481.
Drew TW (1995): Comparative serology of porcine reproductive and respiratory syndrome in eight European laboratories, using immunoperoxidase monolayer assay and enzyme-linked immunosorbent assay. Rev Sci Tech 14: 761–775.
Eclercy J, Renson P, Lebret A, Hirchaud E, Normand V, Andraud M, Paboeuf F, Blanchard Y Rose N, Bourry O (2019): A field recombinant strain derived from two type 1 Porcine Reproductive and Respiratory Syndrome Virus (PRRSV-1) modified live vaccines shows increased viremia and transmission in SPF pigs. Viruses 11: 2916.
Frey T, Richter A, Skrypski J, Schärfe C, Hoferer M (2017): Molecular epidemiology as an effective tool for the diagnosis and control of PRRSV infections in pig farms, illustrated using five cases. Berl Münch Tierärztl Wochenschr 130: 323–331.
Hanada K, Suzuki Y, Nakane T, Hirose O, Gojobori T (2005): The origin and evolution of porcine reproductive and respiratory syndrome viruses. Mol Biol Evol 22: 1024–1031.
Holtkamp DJ, Kliebenstein JB, Neumann EJ, Zimmermann J, Rotto HF, Yoder TK, Wang C, Yeske PE, Mowrer CL, Haley CA (2013): Assessment of the economic impact of porcine reproductive and respiratory syndrome virus on United States pork producers. J Swine Health Prod 21: 72–84.
ICTV (International Committee on Taxonomy of Viruses) (2018): Virus taxonomy 2018b Release. EC 50, Washington, DC, July 2018. https://talk.ictvonline.org.
Jiang H, Lei R, Ding S, Zhu S (2014): Skewer: a fast and accurate adapter trimmer for next-generation sequencing paired-end reads. BMC Bioinformatics 15: 182.
Kappes MA, Faaberg KS (2015): PRRSV structure, replication and recombination: Origin of phenotype and genotype diversity. Virology 479–480: 475–486.
Larochelle R, D’Allaire S, Magar R (2003): Molecular epidemiology of porcine reproductive and respiratory syndrome virus (PRRSV) in Québec. Virus Res 96: 3–14.
Li B, Fang L, Xu Z, Liu S, Gao J, Jiang Y, Chen H, Xiao S (2009): Recombination in Vaccine and Circulating Strains of Porcine Reproductive and Respiratory Syndrome Viruses. Emerg Infect Dis 15: 2032–2035.
Lole KS, Bollinger RC, Paranjape RS, Gadkari D, Kulkarni SS, Novak NG, Ingersoll R, Sheppard HW, Ray SC (1999): Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J Virol 73: 152–160.
Martin DP, Murrell B, Golden M, Khoosal A, Muhire B (2015): RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol 1: vev003.
Martín-Valls GE, Kvisgaard LK, Tello M, Darwich L, Cortey M, Burgara-Estrella AJ, Hernández J, Larsen LE, Mateu E (2014): Analysis of ORF5 and full-length genome sequences of porcine reproductive and respiratory syndrome virus isolates of genotypes 1 and 2 retrieved worldwide provides evidence that recombination Is a common phenomenon and may produce mosaic isolates. J Virol 88: 3170–3181.
Marton S, Szalay D, Kecskeméti S, Forró B, Olasz F, Zádori Z, Szabó I, Molnár T, Bányai K, Bálint Á (2019): Coding-complete sequence of a vaccine-derived recombinant porcine reproductive and respiratory syndrome virus strain isolated in Hungary. Arch Virol 164: 2605–2608.
Mateu E, Díaz I, Darwich L, Casal J, Martín M, Pujols J (2006): Evolution of ORF5 of Spanish porcine reproductive and respiratory syndrome virus strains from 1991 to 2005. Virus Res 115: 198–206.
Montaner-Tarbes S, Del Portillo HA, Montoya M, Fraile L (2019): Key gaps in the knowledge of the porcine respiratory reproductive syndrome virus (PRRSV). Front Vet Sci 6: 38.
Murtaugh MP, Yuan S, Nelson EA, Faaberg KS (2001): Genetic interaction between porcine reproductive and respiratory syndrome virus (PRRSV) strains in cell culture and in animals. J Swine Health Prod 10: 15–21.
Richter A, Frey T, Skrypski J, Hoferer M (2014): Molekulare Epidemiologie als wirksames Instrument für die PRRSV-Diagnostik, Presentation AVID Tagung, Bad Staffelstein.
Pileri E, Mateu E (2016): Review on the transmission porcine reproductive and respiratory syndrome virus between pigs and farms and impact on vaccination. Vet Res 47: 108.
Rathkjen PH, Dall J (2017): Control and eradication of porcine reproductive and respiratory syndrome virus type 2 using a modified-live type 2 vaccine in combination with a load, close, homogenise model: an area elimination study. Acta Vet Scand 59: 4.
Shi M, Lam TT, Hon CC, Hui RK, Faaberg KS, Wennblom T, Murtaugh MP, Stadejek T, Leung FC (2010a): Molecular epidemiology of PRRSV: a phylogenetic perspective. Virus Res 154: 7–17.
Shi M, Lam TT, Hon CC, Murtaugh MP, Davies PR, Hui RK, Li J, Wong LT, Yip CW, Jiang JW, Leung FC (2010b): Phylogeny-based evolutionary, demographical, and geographical dissection of North American type 2 porcine reproductive and respiratory syndrome viruses. J Virol 84: 8700–8711.
Shi M, Lemey P, Singh Brar M, Suchard MA, Murtaugh MP, Carman S, D’Allaire S, Delisle B, Lambert MÈ, Gagnon CA, Ge L, Qu Y, Yoo D, Holmes EC, Chi-Ching Leung F (2013): The spread of type 2 Porcine Reproductive and Respiratory Syndrome Virus (PRRSV) in North America: a phylogeographic approach. Virology 447: 146–154.
Simon-Loriere E, Holmes EC (2011): Why do RNA viruses recombine? Nat Rev Microbiol 9: 617–626.
Stadejek T, Oleksiewicz MB, Potapchuk D, Podgórska K (2006): Porcine reproductive and respiratory syndrome virus strains of exceptional diversity in eastern Europe support the definition of new genetic subtypes. J Gen Virol 87: 1835–1841.
Steinrigl A, Revilla-Fernández S, Schmoll F (2018): Rekombination zwischen PRRSV Feld- und Impfstämmen. 9. Leipziger Tierärztekongress Tagungsband 3, 73–75.
Torrents D, Miranda J, Pedrazuela R, Gauger PC, Ramirez A, Linhares DCL (2019): Implementation of PRRSV status classification system in swine breeding herds from a large integrated group in Spain. Porcine Health Manag 5: 26.
Yuan S, Nelsen CJ, Murtaugh MP, Schmitt BJ, Faaberg KS (1999): Recombination between North American strains of porcine reproductive and respiratory syndrome virus. Virus Res 61: 87–98.
Zhang J, Zheng Y, Xia X-Q, Chen Q, Bade S A, Yoon K-J, Harmon K M, Gauger P C, Main R G, Li G (2017): High-throughput whole genome sequencing of Porcine reproductive and respiratory syndrome virus from cell culture materials and clinical specimens using next-generation sequencing technology. J Vet Diagn Invest 29: 41–50.

Kostenfreier Download
Klicken Sie hier, wenn Sie das PDF BMTW-10.23761439-0299-2020-19-Schneider-Buehl.pdf (0.32 MB) herunterladen möchten
Kostenfreier Download
Klicken Sie hier, wenn Sie das PDF BMTW-10.23761439-0299-2020-19-Schneider-Buehl-Tabelle1.pdf (0.2 MB) herunterladen möchten
Kostenfreier Download
Klicken Sie hier, wenn Sie das PDF BMTW-10.23761439-0299-2020-19-Schneider-Buehl-Tabelle2.pdf (0.2 MB) herunterladen möchten
Kostenfreier Download
Klicken Sie hier, wenn Sie das PDF Supplementary_Table_S1.ods (0.01 MB) herunterladen möchten
Kostenfreier Download
Klicken Sie hier, wenn Sie das PDF Supplementary_Table_S2.ods (3.82 MB) herunterladen möchten
Kostenfreier Download
Klicken Sie hier, wenn Sie das PDF Supplementary_Document_S3.pdf (0.14 MB) herunterladen möchten



{kind=link}
{kind=link}
{kind=link}