

Charakterisierung des Darmmikrobioms in einer Zucht- kolonie von Marmosets
Berliner und Münchener Tierärztliche Wochenschrift 133
DOI: 10.2376/1439-0299-2020-9
© Schlütersche Verlagsgesellschaft mbH & Co. KG. 2020
Publiziert: 8/2020
Summary
Although the microbiome signatures in primates are more similar to humans in comparison to microbiome of mice and rats, the study of microbiome in primate models and particularly in non-human primates such as the common marmoset (Callithrix jacchus) are sparse.
In this study, we analyse the diversity and the taxonomic composition of the gut microbiome of the 13 members of a captive colony of common marmosets located in an experimental facility by high-throughput 16S-rRNA gene sequencing.
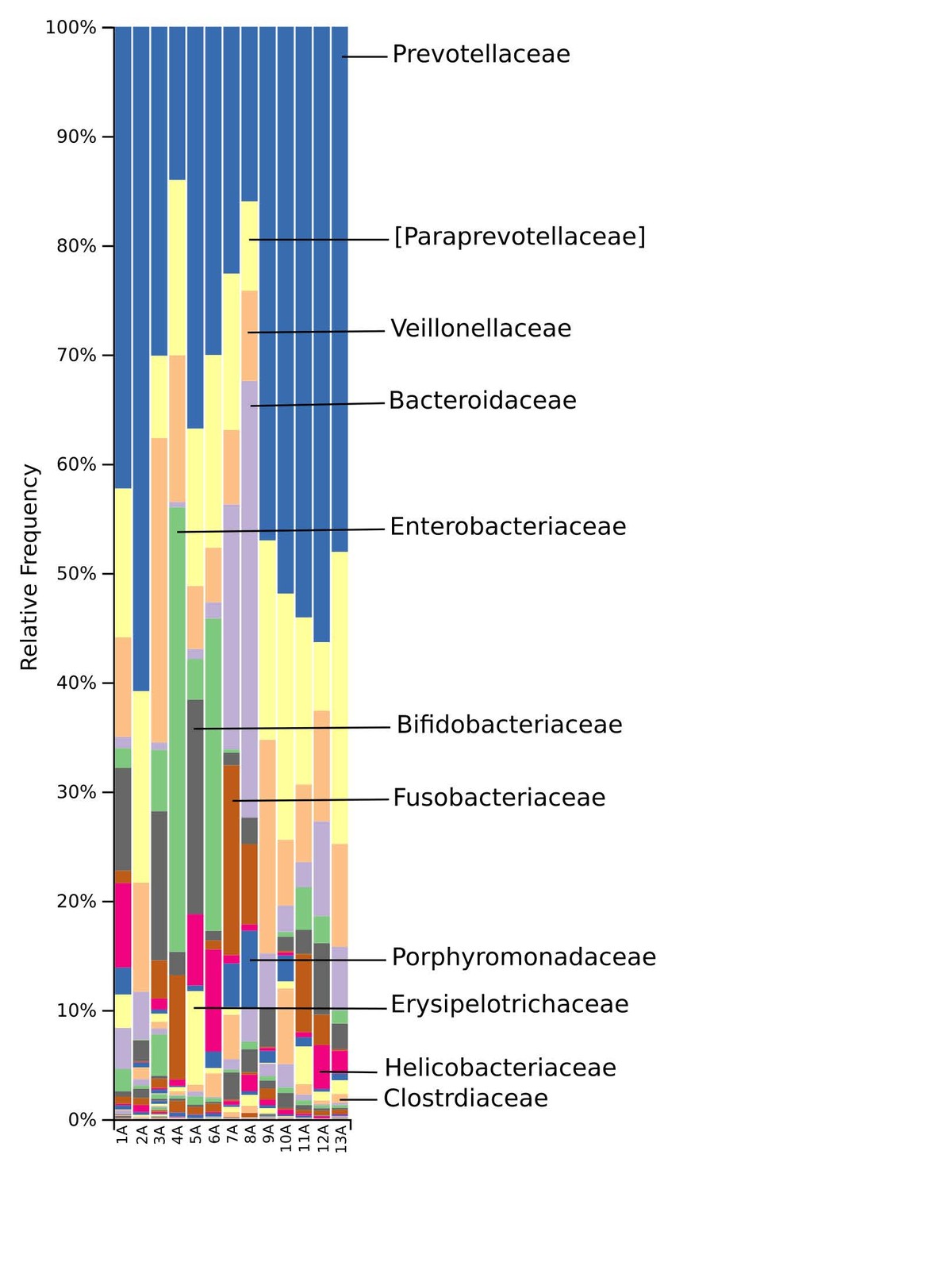
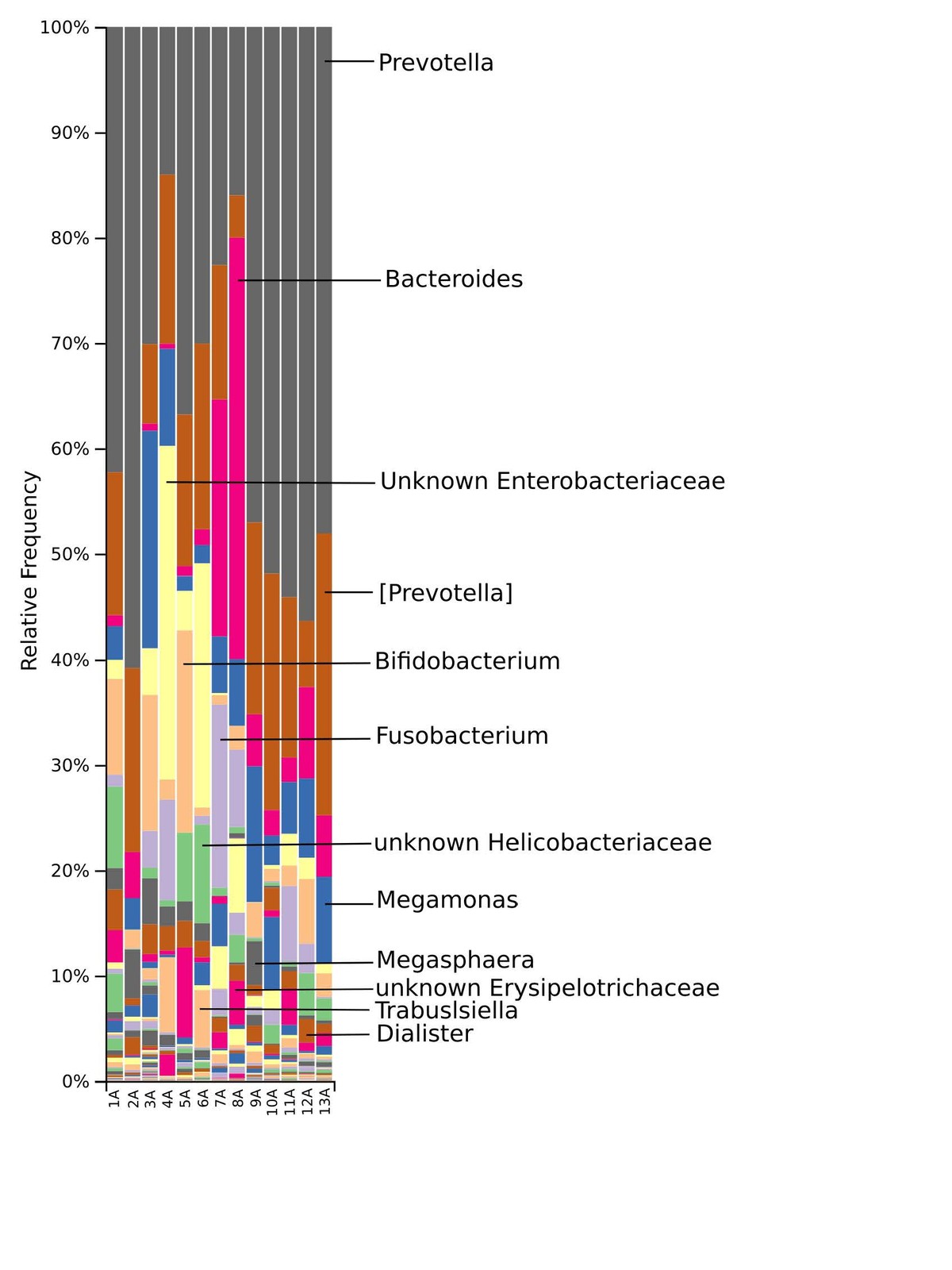
The gut microbiome of the marmosets displayed a complex phylogenetical and taxonomical composition, though similar among the members of the colony. Bacteria of the phyla Bacteroidetes, Firmicutes, Proteobacteria, Fusobacteria and Actinobacteria dominated the baseline microbiota. At family rank Prevotellaceae and Paraprevotellaceae dominate most of the samples, whereas Bacteroidaceae and Enterobacteriaceae were more abundant in some samples. Other families such as Bifidobacteriaceae, Fusobacteriaceae, Porphyromonadaceae, Erysipelotrichaceae, Helicobacteriaceae and Clostridiaceae were recorded in smaller percentages. At genus level, the Prevotella genus was the most abundant in ten of the 13 samples, whereas Bacteroides and an Enterobacteriaceae genus were the most abundant in the remaining samples.
These results supplement and enrich the current knowledge on marmosets gut microbiome as a model for microbiome studies.
Zusammenfassung
Obwohl das Mikrobiom der Primaten im Vergleich zu dem Mikrobiom von Mäusen und Ratten dem Menschen ähnlicher ist, ist die Untersuchung des Mikrobioms in Primatenmodellen und insbesondere in nichtmenschlichen Primaten wie dem Weißbüschelaffen (Callithrix jacchus) spärlich.
In dieser Studie analysieren wir die Diversität und die taxonomische Zusammensetzung des Darmmikrobioms unter den 13 Mitgliedern einer geschlossenen Zuchtkolonie von Weißbüschelaffen, die sich in einer wissenschaftlichen Versuchsanlage befinden, durch 16S-rRNA-Gensequenzierung mit hohem Durchsatz.
Das Darmmikrobiom der Krallenaffen zeigte eine komplexe phylogenetische und taxonomische Zusammensetzung, obwohl sie unter den Mitgliedern der Kolonie ähnlich war. Bakterien der Phyla Bacteroidetes, Firmicutes, Proteobacteria, Fusobacteria und Actinobacteria dominierten die Baseline-Mikrobiota. In den meisten Proben dominieren die Familien Prevotellaceae und Paraprevotellaceae, während Bacteroidaceae und Enterobacteriaceae in einigen Proben häufiger vorkamen. Andere Familien wie Bifidobacteriaceae, Fusobacteriaceae, Porphyromonadaceae, Erysipelotrichaceae, Helicobacteriaceae und Clostridiaceae wurden in kleineren Prozentsätzen aufgezeichnet. Auf Gattungsebene war die Prevotella in zehn der 13 Proben am häufigsten, während die Gattungen Bacteroides und Enterobacteriaceae in den übrigen Proben am häufigsten vorkamen.
Diese Ergebnisse ergänzen und erweitern das aktuelle Wissen über das Darmmikrobiom von Krallenaffen als Modell für Mikrobiomstudien.
Introduction
The gut microbiome represents a complex ecological system with influence on human and animal health and disease (Clemente et al. 2012). Numerous rodent models of human disease, which can be influenced by the composition and diversity of the gut microbiota are available (Hansen et al. 2014). On the contrary, the development of disease models and the study of microbiome in primate models and particularly in non-human primates (NHPs) such as the common marmoset (Callithrix jacchus) are limited (Reveles et al. 2019, Takehara et al. 2019). The microbiome signatures in these animals are more similar to humans in comparison to mice and rats (Nagpal et al. 2018b). Thus, although rodent models have several economical and practical advantages over the primates in the study of host-microbiota interaction, NHPs are necessary to understand this interaction in the context of their much more similar nutrition, environment and pathology (Bauer et al. 2011; Nagpal et al. 2018a, b).
Marmosets are commonly found in laboratory experimental facilities (Winn et al. 2019) and represent a very good model for neuroscience research (Kap et al. 2018) because of their cognitive abilities and suitability for genetic manipulation (Kishi et al. 2014). Due to their smaller size and shorter life-span in comparison to other NHPs, the marmoset constitutes an advantageous primate model for geriatric but also for microbiome studies (Salmon 2016, Takehara et al. 2019). Despite these advantages there are currently just a few studies dealing with the characterization of the microbiota in marmosets (Michelini et al. 2015, Reveles et al. 2019, Shigeno et al. 2018, Takehara et al. 2019).
The goal of the present study was to analyse the diversity and the taxonomic composition of the gut microbiome among all members of the common marmoset colony in our experimental animal facility. We analysed 16S rRNA gene amplicon data, thus focusing on bacterial microbiota.
Top Job:
Material and methods
Animals and sample collection
The sampled marmosets were born in our experimental animal facility. Their maintenance and breeding were approved by the supervisory authority and were carried out under the authorization number 32/12-1-5. All experimental procedures were performed in accordance with the German and European legislation for the care and use of laboratory animals and were conform to institutional standards.
The marmoset colony was housed in a large room, in multiple 250/100/100 (height/width/length) cm individual compartments divided by metal raster. The marmosets lived either in family or in sibling groups, as depicted in Table 1, and had visual contact with their roommates and direct contact with the animals located into the neighbouring compartment through the raster. The living spaces were enriched by various climbing opportunities including hanging ropes, boards and tree branches at different heights. A metal house located at approximately 2 meter height within each compartment served as a retreat and sleeping place. The marmosets were maintained at 27°C, a relative humidity of 70% and on a 12-hour light/dark cycle by using full-spectrum fluorescent tubes that produce not only visible light but also an extra high UV content. Overpressure and a 15-fold air change program were applied in the room.
The marmosets’ nutrition consisted of a changing dietary feeding plan, which included Sniff’s standard commercial pellets V3843-000, Marmoset/New World monkeys 4 mm, with enhanced vitamin D3 content (Sniff, Soest, Germany), baby porridge, rusks, egg, fresh fruit and mealworms. In order to cover the vitamin requirements, the animals also receive D-fluorettes 500 I.E. D3 (Sanofi, Köln, Germany) and Vitacombex (Vitamin A 900.000 I.E., Vitamin D3 72.000 I.E., Vitamin B1 540 mg, Vitamin B2 774 mg, Vitamin B6 180 mg, Vitamin C 9.000 mg, Nicotinamid 3.600 mg, Bentonid 2.500 mg, crude protein 0.9%, crude fat 0.7%, crude fiber 0.1%, raw ash 0.3%) (Quiko, Bochold, Germany). They received ad libitum drinking water from the communal supply and sometimes fennel tea.
For collection of the faecal samples, sterile plastic sheets were placed on the floor of the individual animal compartments during the regular cleaning of the room. The animals were observed discretely until they naturally defecated on the plastic sheets and the faeces were then collected with sterile tweezers into 2 ml sterile cups, which were immediately frozen at –80°C until further use. All samples were collected between 8 and 10 a.m. at the same day.
DNA extraction, 16S amplicon library preparation and sequencing
DNA extraction from faeces was performed using the DNeasy PowerSoil Kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocol. The total DNA was eluted in 100 µl of EB buffer (Qiagen, Hilden, Germany) and frozen at –20°C until further processing. Amplification of 16S rRNA gene V3–V4 regions and library preparation was performed according to the Illumina 16S metagenomics protocol (Part #15044223 Rev. B) using the Nextera XT index kit. Multiplexed sequencing was performed on an Illumina MiSeq machine using v3 chemistry and a 2x300 bp setup. Trimming and demultiplexing of resulting sequence data was performed using standard Illumina procedures.
Informatics and Statistics
Quality controlled sequence data was imported into the Qiita database (https://qiita.ucsd.edu/; hosted at UC San Diego) (Gonzalez et al. 2018) under the study ID 12158. Through Qiita, we used QIIME 1.9.1 (Caporaso et al. 2010) to further control the quality and trim reads to 250 nt. De novo sub-operational taxonomic units (sOTU) were determined using the Deblur 1.1.0 approach (Amir et al. 2017). Taxonomy for Deblur sequences was assigned via the q2-feature-classifier (Bokulich et al. 2018) of Qiime2 version 2018.11 (Bolyen et al. 2019) using the pre-trained Naive Bayes classifier https://data.qiime2.org/2019.1/common/gg-13-8-99-515-806-nb-classifier…, which is based on V4 ribosomal sequences of Greengenes.
Following, the Deblur sOTU table was used to determine the alpha- and beta diversities using Qiime2.
We used q2-fragment-insertion of Qiime2 2018.11 (Janssen et al. 2018) to phylogenetically place all Deblur sequences into the reference Greenegenes 13.8 99% identity tree to obtain a phylogeny for downstream phylogenetic aware alpha- and beta- diversity metrics, i.e. Faith’s phylogenetic diversity index (Faith 1992) and weighted and unweighted UniFrac (Lozupone et al. 2011).
Alpha and Beta Diversity
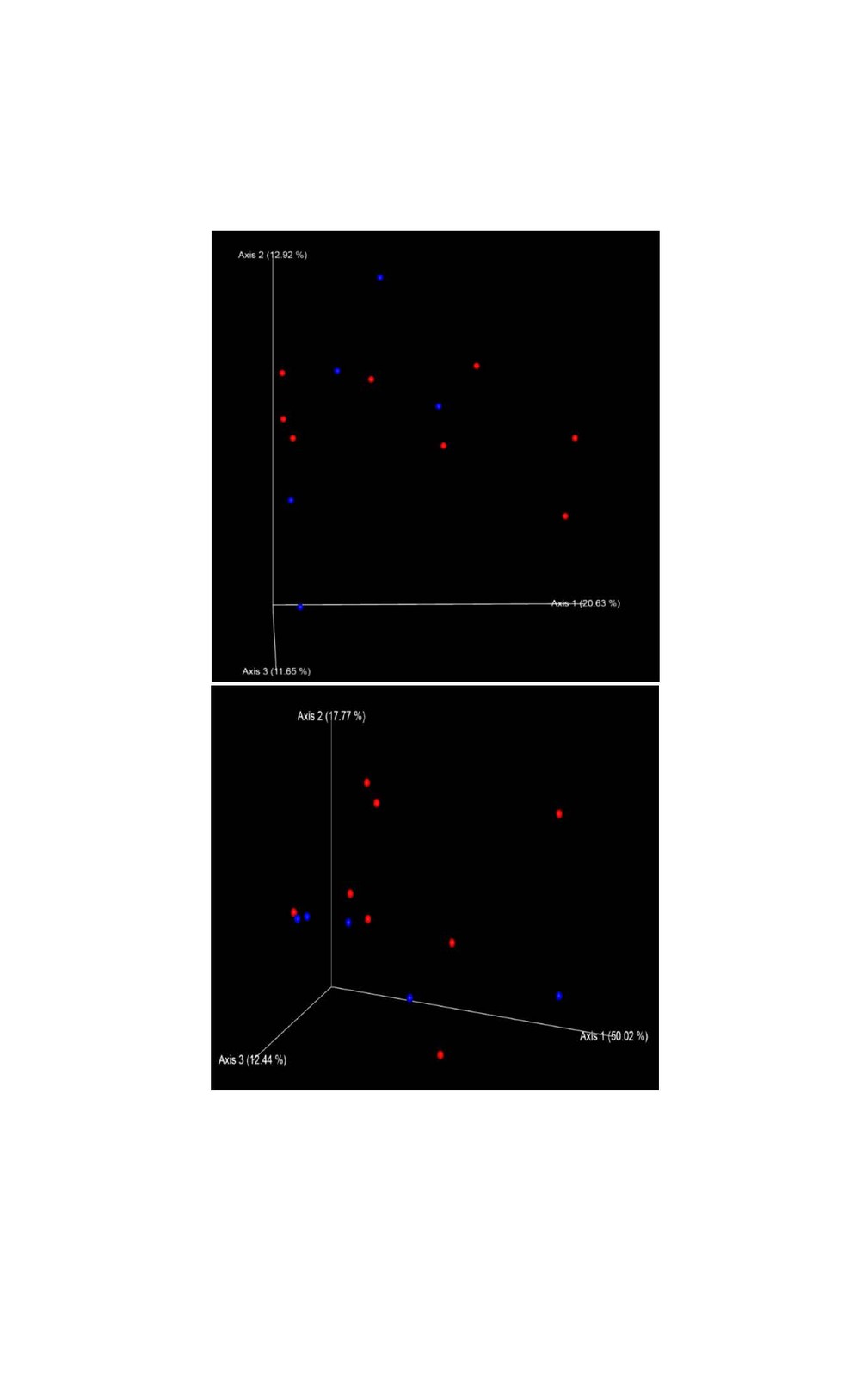
We chose a rarefaction depth of 26,000 reads per sample by analyzing alpha rarefaction curves for the three metrics “observed_OTUs”, “Shannon” and “Faith’s PD” using 10 iterations for every depth. Alpha diversity was calculated using the plain number of observed OTUs (richness), Shannon index and Faith’s phylogenetic diversity index (Faith PD). Beta diversity was defined using the non-parametric phylogenetic measures of weighted and unweighted UniFrac and was visualized as Principal Coordinate Analysis (PCoA) in an 3D Emperor plot (Vazquez-Baeza et al. 2013). All calculations were conducted by applying the Qiime2 program (version 2018.11). The level of significance between the groups of interest were assessed for alpha and beta diversity applying respectively Kruskal-Wallis tests and PERMANOVA using 999 permutations.
Results
Analysis of sequencing results
The total number of sequences per faecal sample processed from the 13 marmosets ranged from 30,355 to 66,194 (55,226 ± 10,461). Alpha rarefaction curves reached a plateau at around 26,000 sequences. We used this value as rarefaction depth for all 13 samples.
Faeces microbiome diversity
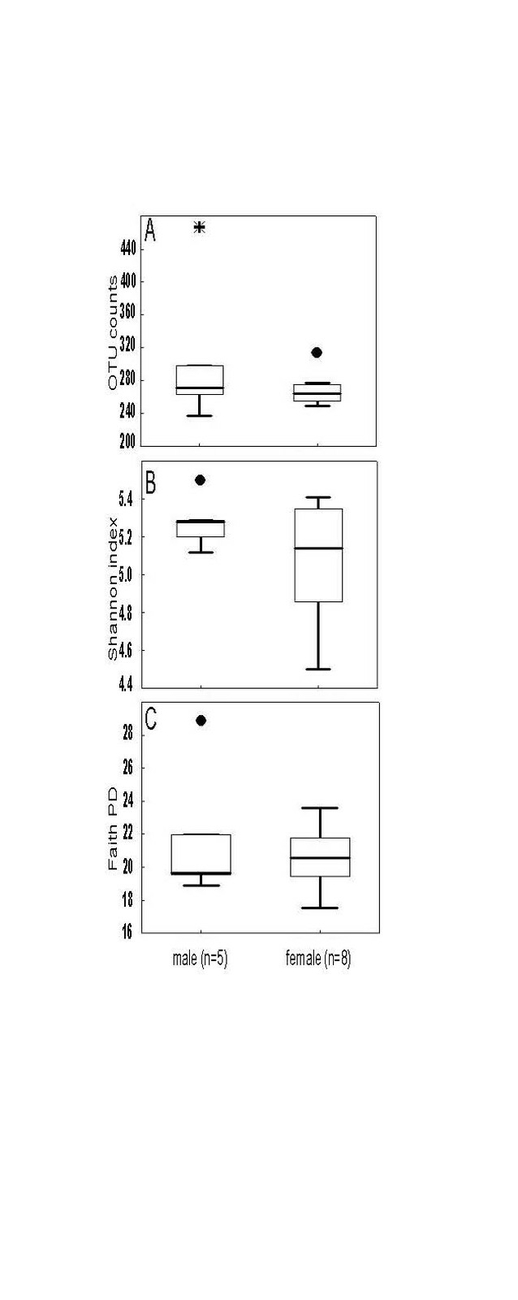
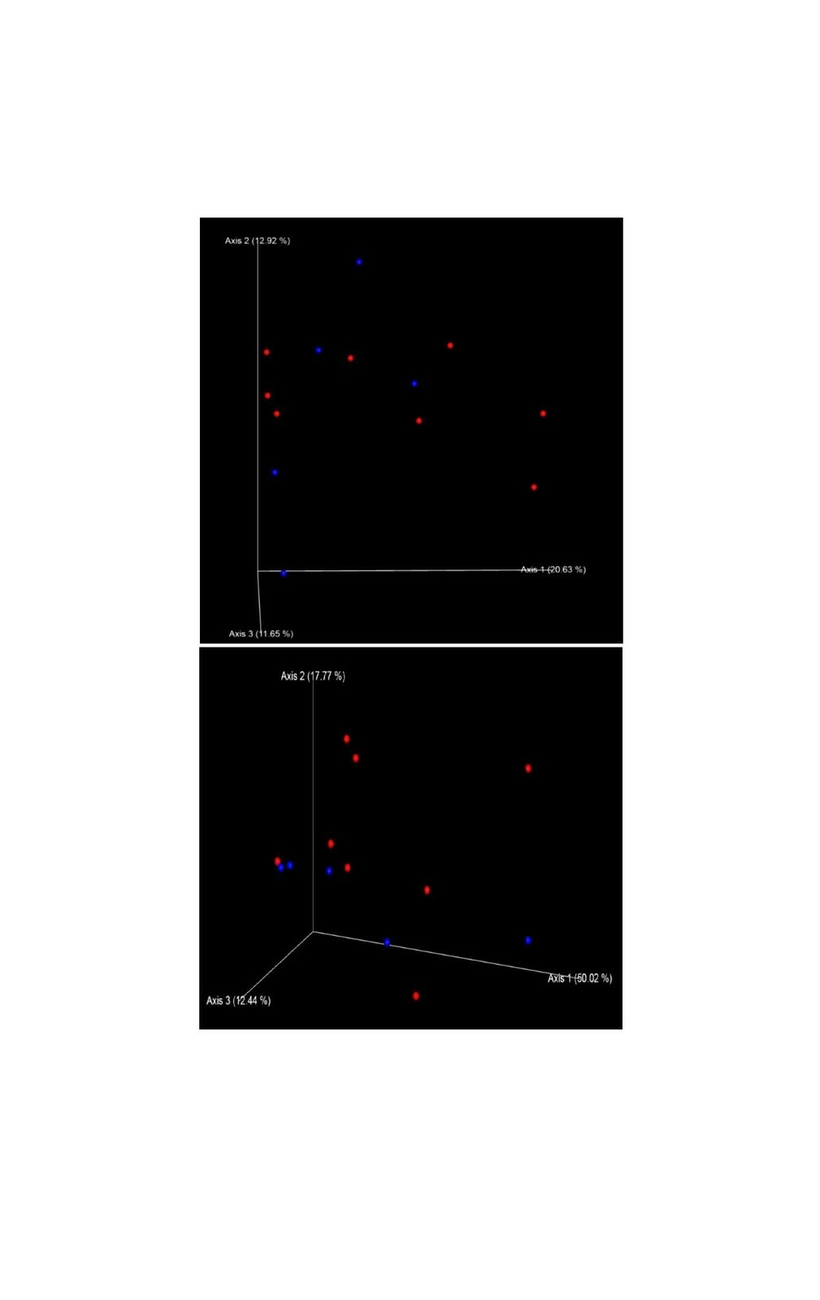
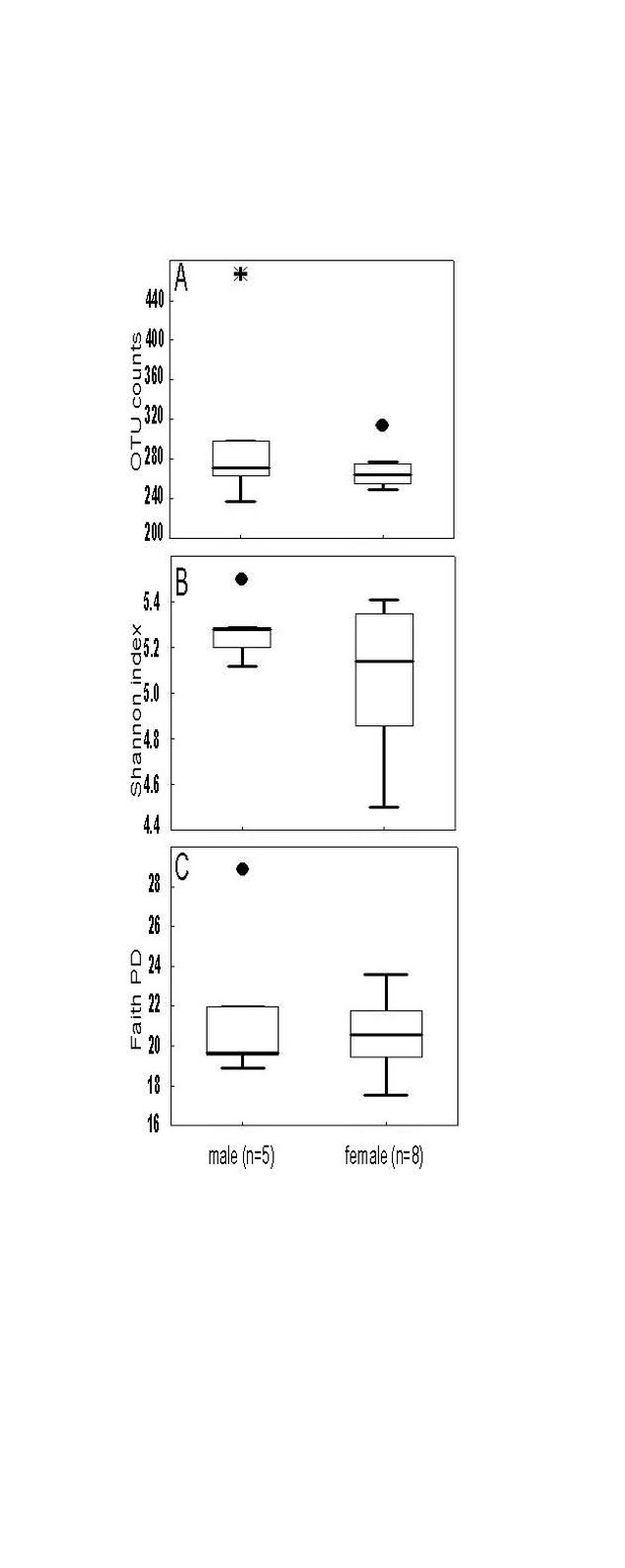
Alpha diversity metrics richness (number of observed OTUs), Shannon index and Faith’s PD were employed to characterize the phylogenetic diversity of the samples separately for each gender. No significant differences between the sexes were found (Fig. 1). The richness among the 13 samples ranged from 236.00 to 466.00 (283.00 ± 56.00) OTUs, the Shannon index varied from 4.50 to 5.50 (5.15 ± 0.26), whereas the Faith’s PD values were between 17.50 and 28.80 (21.00 ± 2.72). To analyse gender differences in the bacterial communities, unweighted and weighted UniFrac distances were computed and visualized as 3D Principal Coordinate Analysis (PCoA) plots (Fig. 2). No statistically significant gender differences could be noticed, which indicates similar presence and abundance of taxa within the samples.
Baseline taxonomical composition of fecal microbiota
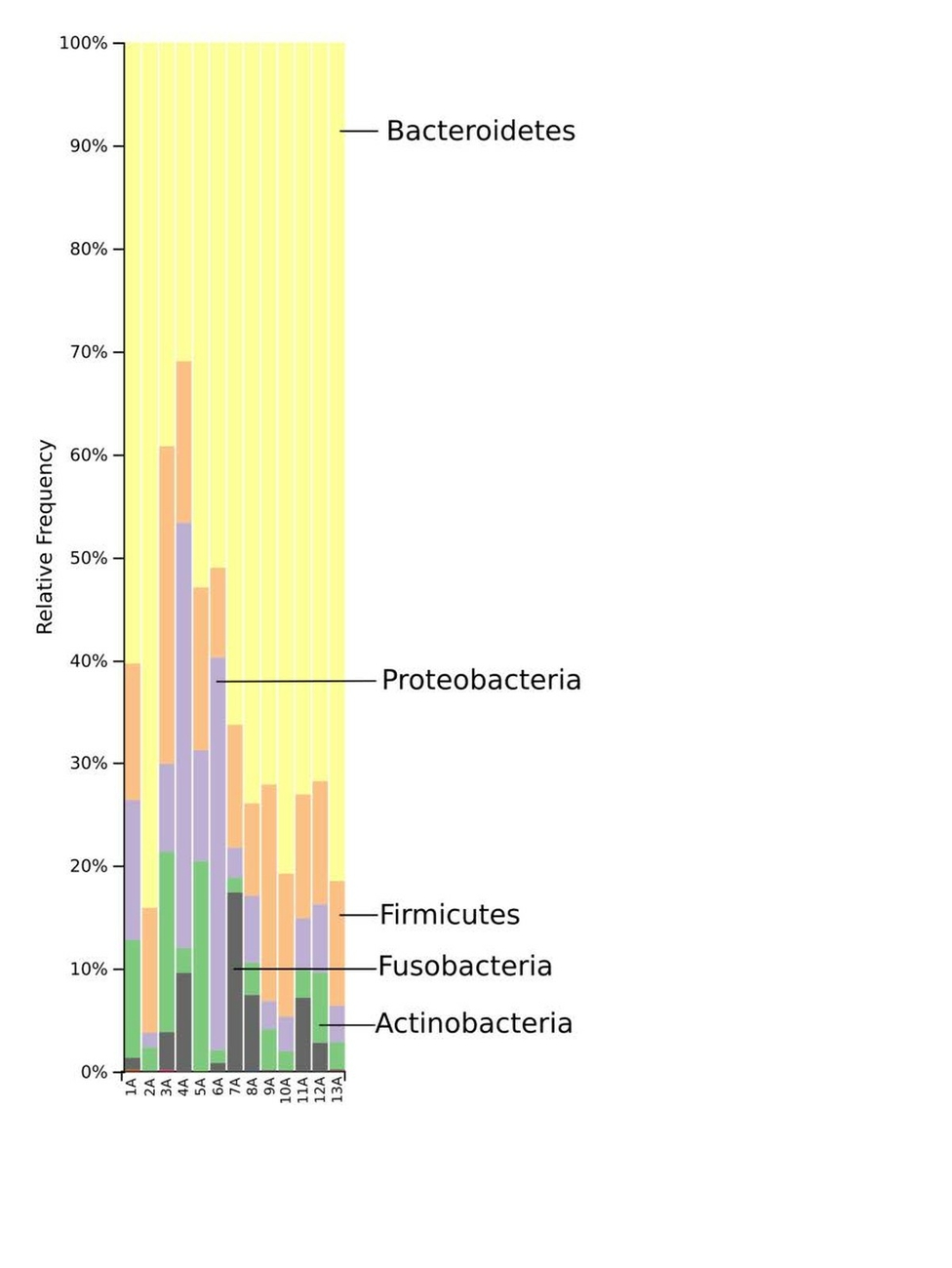
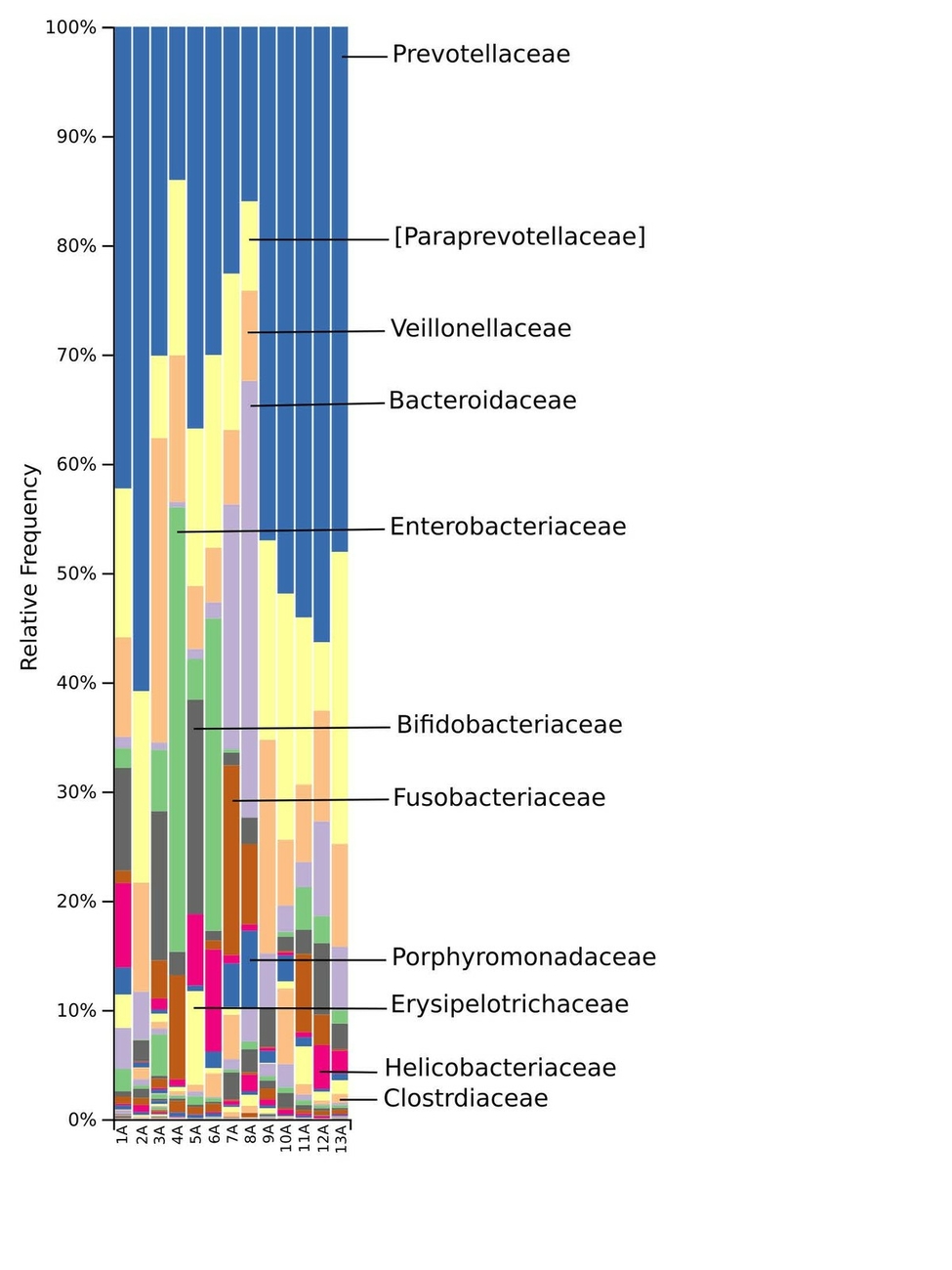
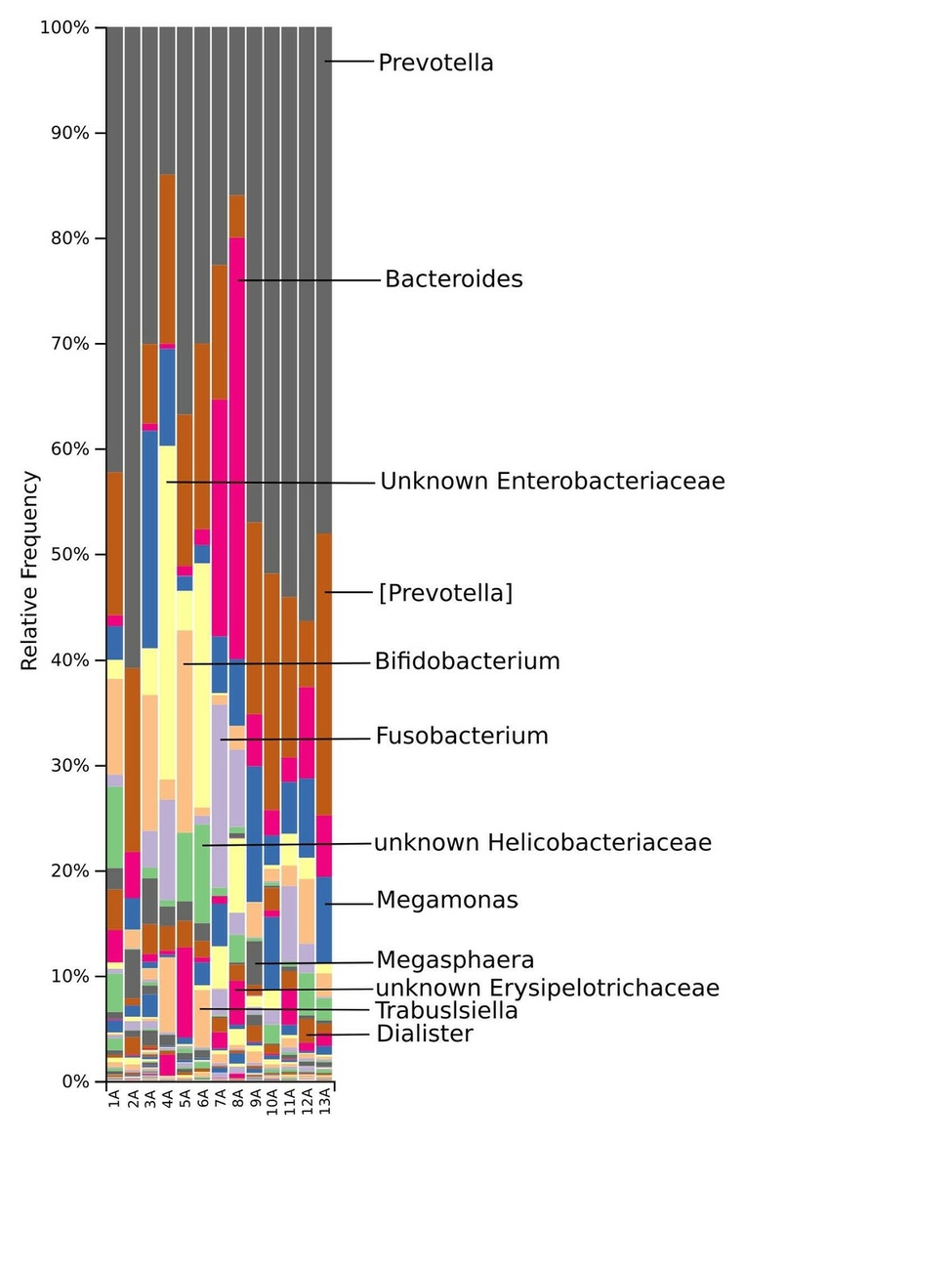
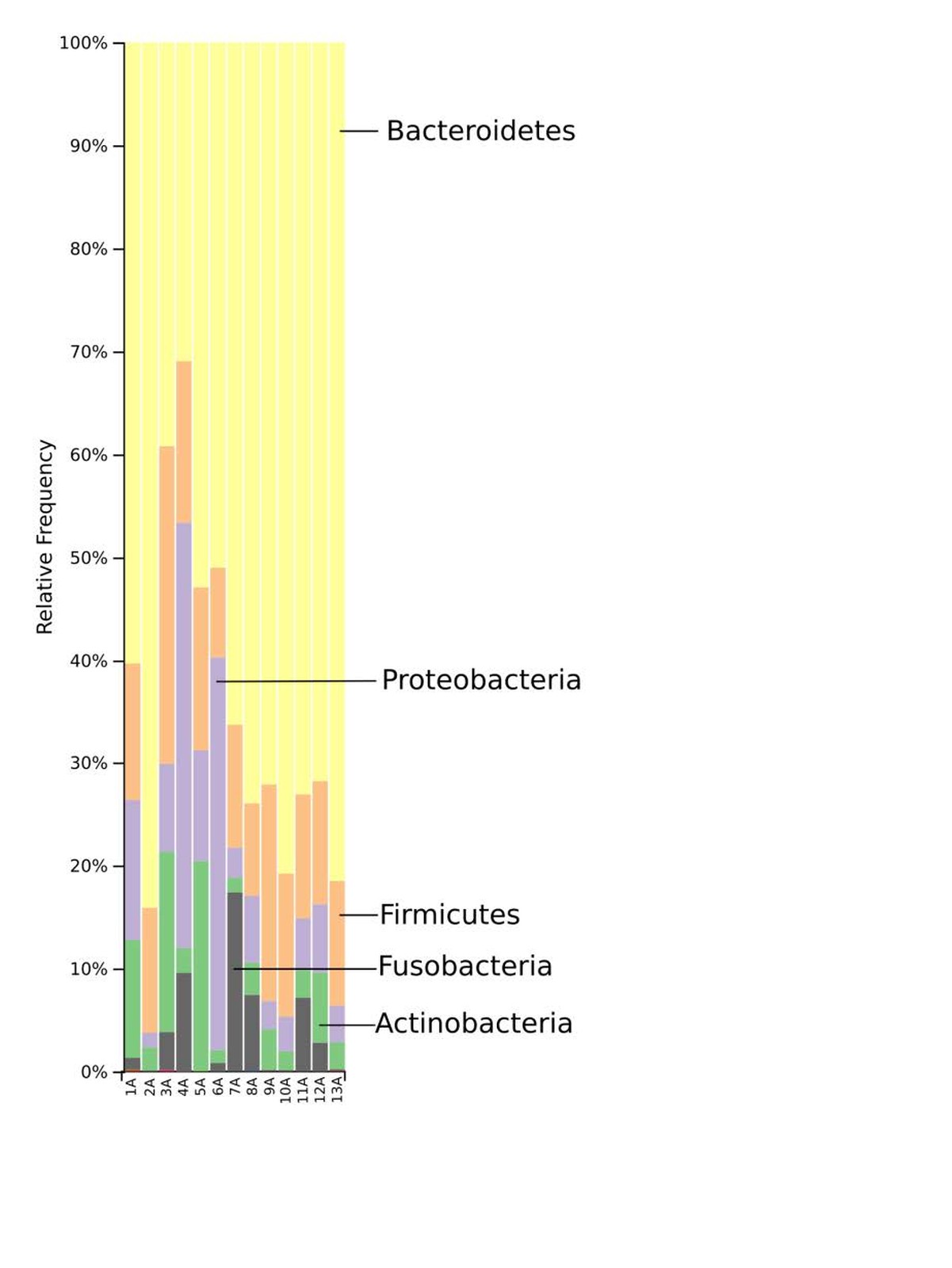
The taxonomic distribution derived from the 16S rRNA gene sequences was analysed from phyla to genus level. Bacteria of the phyla Bacteroidetes, Firmicutes and Proteobacteria dominated the baseline microbiota. In addition, high abundance of the Fusobacteria and Actinobacteria was recorded in nearly all samples (Fig. 3). At family level, Prevotellaceae and Paraprevotellaceae dominated most of the samples, whereas Bacteroidaceae and Enterobacteriaceae were dominant in only some of the samples. Other families such as Bifidobacteriaceae, Fusobacteriaceae, Porphyromonadaceae, Erysipelotrichaceae, Helicobacteriaceae and Clostridiaceae were present in smaller percentages among the microbiomes of the 13 marmosets (Fig. 4). The Prevotella genus was with 30–60% relative abundance the most abundant in ten of the 13 samples. The most abundant genera in marmoset 7 were Bacteroides and Prevotella each counting for 22%, whereas Bacteroides and a genus belonging to Enterobacteriaceae were with 40% and 31% the most abundant genera in the samples 8 and 4 respectively (Fig. 5). Interestingly, the gut microbiome of the twin marmosets 7 and 8, 10 and 11, 12 and 13 that are housed together, display quite similar patterns within the twin pair.
Discussion
The study of the gut microbiome in marmosets is still in its incipient stage. In the current work, we analysed the faecal microbiome of all members of a captive family of common marmosets. Our findings supplement and expand the actual knowledge on the microbiome of this non-human primate species. The richness values recorded here (Fig. 1A) were similar with those from a previous study (Reveles et al. 2019), to our knowledge the only study analysing the gut microbiome in marmosets by next-generation sequencing. The Shannon index of the present study (Fig. 1B) was higher in comparison to Reveles et al. (2019). However, differences are to be expected when populations located in different environments are compared. The most evident effect of the environment should be expected when comparing captive vs. wild living animals. Wild Guizhou snub-nosed monkey (Rhinopithecus brelichi) for example exhibited increased gut microbial diversity in comparison with captive monkey based on the Chao1 but not Shannon diversity metric (Hale et al. 2019). Reduced microbial diversity in captivity in black howler monkey (Alouatta pigra) has been associated for example with lower dietary diversity (Amato et al. 2013).
The phylogenetic diversity index Faith’s PD with values of around 21 displayed a relative diverse microbial composition, similar among the members of the colony (Fig. 1C). The similarity within the gut microbiota between the 13 members of the colony was further analysed by the beta diversity metrics unweighted and weighted UniFrac (Fig. 2), which, however, did not show clear differences between the samples analysed. As far as the gender of the marmoset population is concerned, no statistically significant differences in the gut microbiome were found. The broad age distribution and the number of marmosets available in our colony did not allow an age related statistical analysis of the microbiome. Nevertheless, one can notice a shift in the taxonomy pattern of the microbiome from the oldest individual (sample 1) to the youngest (sample 13) (Fig. 3, 4, 5), suggesting an age related change of the diversity and composition of the gut microbiome from geriatric to young adult marmosets, as recently documented (Reveles et al. 2019). The baseline microbiota was dominated by bacteria of the phyla Bacteroidetes, Firmicutes, Proteobacteria, Actinobacteria and Fusobacteria (Fig. 3), which is in accordance with previous reports from studies on humans and other non-human primates (Chen et al. 2018). Nevertheless, Bacteroidetes dominated the microbiota at phyla level regardless of age and sex. Changes in the Firmicutes/Bacteroidetes ratio seem to correlate with serious pathologies such as obesity and type 2 diabetes in humans (Grigorescu and Dumitrascu 2016). At genus rank, Prevotella was the most abundant in most of the samples, followed by Bacteroides and an unknown genera belonging to Enterobacteriaceae. Interestingly, bacteria belonging to Bifidobacterium were noticed in the faecal samples of the marmosets tested. Several Bifidobacterium species have been previously isolated and cultivated from marmosets (Endo et al. 2012, Michelini et al. 2016, Modesto et al. 2018, 2019). Members of the genus Bifidobacterium are supposed to confer health-promoting effects to their hosts by contributing to the balance of the gut microbiota and modulating the immune system (Milani et al. 2015). Environmental differences lead usually to variations in the abundance of particular taxa. For exemple wild-living snub-nosed monkeys display greater relative abundances of bacteria in the Lachnospiraceae and Ruminococcaceae families than captive monkeys, which conversely had greater relative abundances of Prevotella and Bacteroides species, which degrade simple sugars and carbohydrates, present in the captive snub-nosed monkeys diet (Hale et al. 2019). Overall, the gut microbiome diversity and composition recorded in this study suggest that the common maternal legacy and the environment essentially contribute in the shaping of gut microbiota, since the microbiome composition of individuals housed together and descending from the same mother were the most similar.
In summary, the gut microbiome of the marmosets analysed in this study seems to display a complex phylogenetical and taxonomical composition, similar among the members of the colony. These results supplement and expand the current knowledge on gut microbiome in marmosets, thus contributing to the characterisation of the marmoset as a model for further experimental microbiome studies. Multiple experimental settings in marmosets, for example in the study of aging (Reveles et al. 2019), multiple sclerosis (Kap et al. 2018), oral diseases (Takehara et al. 2019), infectious diseases (Yamazaki et al. 2017), imply the microbiome. Further experimentation is required to reveal the mechanisms and factors that create, maintain and influence the microbiome in order to transfer the knowledge to human level.
Acknowledgements
We gratefully acknowledge Sandra Plante for the excellent technical assistance.
Conflict of interest
The authors declare that they have no competing interests.
Funding
Not applicable.
Ethical approval
Not applicable.
Adress for correspondence
Laurentiu Benga
ZETT – Zentrale Einrichtung für Tierforschung und wiss. Tierschutzaufgaben
Universitätsklinikum/Heinrich-Heine-Universität
Universitätsstr. 1
40225 Düsseldorf
Laurentiu.Benga@med.uni-duesseldorf.de
References
Amato KR, Yeoman CJ, Kent A, Righini N, Carbonero F, Estrada A, Leigh SR (2013): Habitat degradation impacts black howler monkey (Alouatta pigra) gastrointestinal microbiomes. ISME Journal 7: 1344–1353.
Amir A, McDonald D, Navas-Molina JA, Kopylova E, Morton JT, Zech Xu Z, Kightley EP, Thompson LR, Hyde ER, Gonzalez A, Knight R (2017): Deblur Rapidly Resolves Single-Nucleotide Community Sequence Patterns. mSystems 2.
Bauer SA, Arndt TP, Leslie KE, Pearl DL, Turne PV (2011): Obesity in rhesus and cynomolgus macaques: a comparative review of the condition and its implications for research. Comp Med 61: 514–526.
Bokulich NA, Kaehler BD, Rideout JR, Dillon M, Bolyen E, Knight R, Huttley GA, Gregory Caporaso J (2018): Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 6: 90.
Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, Alexander H, Alm EJ, Arumugam M, Asnicar F, Bai Y, Bisanz JE, Bittinger K, Brejnrod A, Brislawn CJ, Brown CT, Callahan BJ, Caraballo-Rodriguez AM, Chase J, Cope EK, Da Silva R, Diener C, Dorrestein PC, Douglas GM, Durall DM, Duvallet C, Edwardson CF, Ernst M, Estaki M, Fouquier J, Gauglitz JM, Gibbons SM, Gibson DL, Gonzalez A, Gorlick K, Guo J, Hillmann B, Holmes S, Holste H, Huttenhower C, Huttley GA, Janssen S, Jarmusch AK, Jiang L, Kaehler BD, Kang KB, Keefe CR, Keim P, Kelley ST, Knights D, Koester I, Kosciolek T, Kreps J, Langille MGI, Lee J, Ley R, Liu YX, Loftfield E, Lozupone C, Maher M, Marotz C, Martin BD, McDonald D, McIver LJ, Melnik AV, Metcalf JL, Morgan SC, Morton JT, Naimey AT, Navas-Molina JA, Nothias LF, Orchanian SB, Pearson T, Peoples SL, Petras D, Preuss ML, Pruesse E, Rasmussen LB, Rivers A, Robeson MS, 2nd Rosenthal P, Segata N, Shaffer M, Shiffer A, Sinha R, Song SJ, Spear JR, Swafford AD, Thompson LR, Torres PJ, Trinh P, Tripathi A, Turnbaugh PJ, Ul-Hasan S, van der Hooft JJJ, Vargas F, Vazquez-Baeza Y, Vogtmann E, von Hippel M, Walters W, Wan Y, Wang M, Warren J, Weber KC, Williamson CHD, Willis AD, Xu ZZ, Zaneveld JR, Zhang Y, Zhu Q, Knight R, Caporaso JG (2019): Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol 37: 852–857.
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R (2010): QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7: 335–336.
Chen Z, Yeoh YK, Hui M, Wong PY, Chan MCW, Ip M, Yu J, Burk RD, Chan FKL, Chan PKS (2018): Diversity of macaque microbiota compared to the human counterparts. Sci Rep 8: 15573.
Clemente JC, Ursell LK, Parfrey LW, Knight R (2012): The impact of the gut microbiota on human health: an integrative view. Cell 148: 1258–1270.
Endo A, Futagawa-Endo Y, Schumann P, Pukall R, Dicks LM (2012): Bifidobacterium reuteri sp. nov., Bifidobacterium callitrichos sp. nov., Bifidobacterium saguini sp. nov., Bifidobacterium stellenboschense sp. nov. and Bifidobacterium biavatii sp. nov. isolated from faeces of common marmoset (Callithrix jacchus) and red-handed tamarin (Saguinus midas). Syst Appl Microbiol 35: 92–97.
Faith DP (1992): Conservation evaluation and phylogenetic diversity. Biol Conservat 61: 1–10.
Gonzalez A, Navas-Molina JA, Kosciolek T, McDonald D, Vazquez-Baeza Y, Ackermann G, DeReus J, Janssen S, Swafford AD, Orchanian SB, Sanders JG, Shorenstein J, Holste H, Petrus S, Robbins-Pianka A, Brislawn CJ, Wang M, Rideout JR, Bolyen E, Dillon M, Caporaso JG, Dorrestein PC, Knight R (2018): Qiita: rapid, web-enabled microbiome meta-analysis. Nat Methods 15: 796–798.
Grigorescu I, Dumitrascu DL (2016): Implication of gut microbiota in diabetes mellitus and obesity. Acta Endocrinol (Buchar) 12: 206–214.
Hale VL, Tan CL, Niu K, Yang Y, Zhang Q, Knight R, Amato KR (2019): Gut microbiota in wild and captive Guizhou snub-nosed monkeys, Rhinopithecus brelichi. Am J Primatol 81: e22989.
Hansen AK, Hansen CH, Krych L, Nielsen DS (2014): Impact of the gut microbiota on rodent models of human disease. World J Gastroenterol 20: 17727–17736.
Janssen S, McDonald D, Gonzalez A, Navas-Molina JA, Jiang L, Xu ZZ, Winker K, Kado DM, Orwoll E, Manary M, Mirarab S, Knight R (2018): Phylogenetic Placement of Exact Amplicon Sequences Improves Associations with Clinical Information. mSystems 3.
Kap YS, Bus-Spoor C, van Driel N, Dubbelaar ML, Grit C, Kooistra SM, Fagrouch ZC, Verschoor EJ, Bauer J, Eggen BJL, Harmsen HJM, Laman JD, t Hart BA (2018): Targeted Diet Modification Reduces Multiple Sclerosis-like Disease in Adult Marmoset Monkeys from an Outbred Colony. J Immunol 201: 3229–3243.
Kishi N, Sato K, Sasaki E, Okano H (2014): Common marmoset as a new model animal for neuroscience research and genome editing technology. Dev Growth Differ 56: 53–62.
Lozupone C, Lladser ME, Knights D, Stombaugh J, Knight R (2011): UniFrac: an effective distance metric for microbial community comparison. ISME J 5: 169–172.
Michelini S, Modesto M, Oki K, Stenico V, Stefanini I, Biavati B, Watanabe K, Ferrara A, Mattarelli P (2015): Isolation and identification of cultivable Bifidobacterium spp. from the faeces of 5 baby common marmosets (Callithrix jacchus L.). Anaerobe 33: 101-104.
Michelini S, Oki K, Yanokura E, Shimakawa Y, Modesto M, Mattarelli P, Biavati B, Watanabe K (2016): Bifidobacterium myosotis sp. nov., Bifidobacterium tissieri sp. nov. and Bifidobacterium hapali sp. nov., isolated from faeces of baby common marmosets (Callithrix jacchus L.). Int J Syst Evol Microbiol 66: 255–265.
Milani C, Lugli GA, Duranti S, Turroni F, Mancabelli L, Ferrario C, Mangifesta M, Hevia A, Viappiani A, Scholz M, Arioli S, Sanchez B, Lane J, Ward DV, Hickey R, Mora D, Segata N, Margolles A, van Sinderen D, Ventura M (2015): Bifidobacteria exhibit social behavior through carbohydrate resource sharing in the gut. Sci Rep 5: 15782.
Modesto M, Michelini S, Oki K, Biavati B, Watanabe K, Mattarelli P (2018): Bifidobacterium catulorum sp. nov., a novel taxon from the faeces of the baby common marmoset (Callithrix jacchus). Int J Syst Evol Microbiol 68: 575–581.
Modesto M, Watanabe K, Arita M, Satti M, Oki K, Sciavilla P, Patavino C, Camma C, Michelini S, Sgorbati B, Mattarelli P (2019): Bifidobacterium jacchi sp. nov., isolated from the faeces of a baby common marmoset (Callithrix jacchus). Int J Syst Evol Microbiol 69: 2477–2485.
Nagpal R, Shively CA, Appt SA, Register TC, Michalson KT, Vitolins MZ, Yadav H (2018a): Gut Microbiome Composition in Non-human Primates Consuming a Western or Mediterranean Diet. Front Nutr 5: 28.
Nagpal R, Wang S, Solberg Woods LC, Seshie O, Chung ST, Shively CA, Register TC, Craft S, McClain DA, Yadav H (2018b): Comparative Microbiome Signatures and Short-Chain Fatty Acids in Mouse, Rat, Non-human Primate, and Human Feces. Front Microbiol 9: 2897.
Reveles KR, Patel S, Forney L, Ross CN (2019): Age-related changes in the marmoset gut microbiome. Am J Primatol 81: e22960.
Salmon AB (2016): Moving toward ‘common’ use of the marmoset as a non-human primate aging model. Pathobiol Aging Age Relat Dis 6: 32758.
Shigeno Y, Toyama M, Nakamura M, Niimi K, Takahashi E, Benno Y (2018): Comparison of gut microbiota composition between laboratory-bred marmosets (Callithrix jacchus) with chronic diarrhea and healthy animals using terminal restriction fragment length polymorphism analysis. Microbiol Immunol 62: 702–710.
Takehara S, Zeredo JL, Kumei Y, Kagiyama K, Fukasawa K, Oshiro A, Ueno M, Kojimahara N, Minakuchi S, Kawaguchi Y (2019): Characterization of oral microbiota in marmosets: Feasibility of using the marmoset as a human oral disease model. PLoS One 14: e0207560.
Vazquez-Baeza Y, Pirrung M, Gonzalez A, Knight R (2013): EMPeror: a tool for visualizing high-throughput microbial community data. Gigascience 2: 16.
Winn CB, Issa EB, Curcillo CP, Townes CA, Burns MA, Patterson MM (2019): Daily Water Intake by Common Marmosets (Callithrix jacchus) and Recommendations Regarding Fluid Regulation. J Am Assoc Lab Anim Sci 58: 16–20.
Yamazaki Y, Kawarai S, Morita H, Kikusui T, Iriki A (2017): Faecal transplantation for the treatment of Clostridium difficile infection in a marmoset. BMC Vet Res 13: 150.

Kostenfreier Download
Klicken Sie hier, wenn Sie das PDF BMTW-10.23761439-0299-2020-9-Benga.pdf (0.28 MB) herunterladen möchten
Kostenfreier Download
Klicken Sie hier, wenn Sie das PDF Tabelle-1-BMTW-10.23761439-0299-2020-9-Benga.pdf (0.12 MB) herunterladen möchten




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}